
Abstract
Fibroblast growth factor 7 (FGF7) is a member of the fibroblast growth factor (FGF) family of proteins. FGF7 is of stromal origin and produces a paracrine effect on epithelial cells. In the current investigation, we aimed to identify new single-domain antibodies (sdAbs) against FGF7 using phage display technology. The vector harboring the codon-optimized DNA sequence for FGF7 protein was transformed into Escherichia coli BL21 (DE3) pLysS, and then the protein was expressed at the optimized condition. Enzyme-linked immunosorbent assay, circular dichroism spectropolarimetry, and in vitro scratch assay experiments were used to confirm the proper folding and functionality of the purified FGF7 protein. The purity of the produced FGF7 was 92%, with production yield of 3.5 mg/L of culture. Panning against the purified FGF7 was performed, and the identified single-domain antibodies showed significant affinity. Further investigation on one of the selected sdAb displaying phage clones showed concentration-dependent binding to FGF7. The selected sdAb can be used for developing novel tumor-suppressing agents where inhibition of FGF7 is required.
Introduction
Fibroblast growth factor 7 (FGF7) or keratinocyte growth factor (KGF) is a member of the fibroblast growth factor (FGF) family of proteins. FGF7 is of stromal origin and produces a paracrine effect on epithelial cells. 1 FGF7 plays versatile roles in proliferation and differentiation on a wide range of epithelial cells like goblet cells, hepatocytes, pulmonary type 2 pneumocytes, mammary gland epithelial cells, pancreatic ductal epithelial cells, and translational urothelial cells. This protein is also involved in the healing process, which makes it an agent for reducing inflammation and injury.2,3 For instance, Palifermin® is a truncated form of FGF7 available on the market for controlling oral mucositis occurring due to adverse effects during chemotherapy and radiotherapy. 2 From a structural point of view, FGF7, with a molecular weight of 19 kDa, contains 194 amino acids that folds into 10 β strands forming five antiparallel β sheets.1,4 Its first 31 N-terminal residues form the signal peptide. This protein contains two disulfide bonds linking cysteines 32 to 46 and 133 to 137. 5
There are several pieces of evidence indicating the contribution of FGF7 in tumorigenesis. The expression and activity profile of FGF7 have been investigated in many cancers such as prostate, breast, colorectal, and lung, demonstrating its pivotal role in proliferation and regulation of these conditions.6–8
It is well accepted that tumors are induced by genetic alterations. Yet complex interactions between extracellular matrix (ECM), stromal and epithelial components in the microenvironment of tumors play an important role in the progression of cancer. 9 Fibroblasts that are involved in the development of some cancers have been investigated as one of the important components of stroma in tumor tissues. 10 These cells, so-called carcinoma-associated fibroblasts (CAFs), in the tumor environment are different from normal fibroblasts in terms of biological and morphological aspects. Signaling and communication between CAFs and different compartments of tumor tissue are mediated through growth factors like FGFs, endothelial growth factor (EGF), and vascular endothelial growth factor (VEGF), leading to angiogenesis and differentiation of tumor cells. As a consequence, CAFs and associated signaling proteins have been introduced as novel targets for cancer therapy.11–13
Phage display technology, introduced by Smith in 1985, 14 is a combinatorial biology approach applicable for the identification of novel antibodies against targets of interest with the promise of resolving difficulties intrinsic to traditional methods. There are numerous reports in which this technology has been used for different purposes.15–17 Several investigations based on phage display have been conducted to study protein/DNA-protein interactions and epitope mapping; to identify organ-, cell-, and disease-specific peptides; and to develop peptide- and protein-mediated drug delivery and molecular imaging systems.18–20
The antigen binding property of antibodies can be retained almost with the same specificity by their fragments carrying the variable domains. Different fragmented forms of antibodies have been proved useful in a variety of applications. Expression level of fragment antibodies in prokaryotic cells surpasses that of full-length antibodies as a result of their smaller size and altered physicochemical properties. Lower cost of production, less immunogenicity, less side effects, and easy penetration into tissues as a result of smaller size have made these proteins promising candidates for therapeutic applications. 21 Single-chain variable fragments (scFvs) and single-domain antibodies are among the most frequently used fragments that can be expressed on the phage surface as a fusion protein attached to one of the coat proteins and hence can be exploited in high-throughput studies. Single-domain antibodies (sdAbs, also known as nanobodies) were initially introduced by Ward et al. 22 and are isolated single heavy chain variable domains (VH) that can demonstrate good binding affinity to variable targets.
Recently, selection of novel fragment antibodies by means of phage display against target proteins and receptors involved in cancer progression has gained great attention.23,24 In this framework, several attempts have been conducted to select such antibodies against fibroblast growth factor receptors (FGFRs).25,26 The aim of the current investigation was to identify domain antibodies against FGF7, an important mediator of CAFs, using phage display technology. The identified domain antibodies can be used in drug design and development processes where inhibition of FGF7 is required.
Materials and Methods
Reagents
All of the chemicals used in this work were of biological grade. Tryptone, agar, and glycerol were obtained from Applichem (Darmstadt, Germany). NaCl and polyethylene glycol (PEG) 6000 were purchased from Scharlau (Barcelona, Spain). Primers used in this work were supplied from Bioron (Ludwigshafen, Germany) ordered via FAZA Biotech (Tehran, Iran). Gel purification and plasmid mini extraction kits were obtained from Bioneer (Daejeon, South Korea). A domain antibody (dAb) library kit was purchased from MRC HGMP Resource Centre. Anti M13–horseradish peroxidase (HRP)–conjugated monoclonal antibody was prepared from Sino Biological (Beijing, China). TMB (3,3′,5,5′-tetramethylbenzidine) was obtained from Sigma-Aldrich (St. Louis, MO). Agarose was from Invitrogen (Paisley, UK). Sodium azide (NaN3) and methanol were from Merck (Darmstadt, Germany). A PCR master kit was purchased from CinnaGen (Tehran, Iran). Nickel sepharose 6 fast flow was prepared from GE Healthcare Life Sciences (Sweden). FGF-7 antibody (A-9) sc-515014 and goat anti-mouse IgG antibody HRP conjugate were from Santa Cruz Biotechnology, and anti-His antibody was from GE Healthcare/Sigma-Aldrich. The BM Chemiluminescence Western blotting kit was obtained from Roche (Berlin, Germany). The MCF-7 cell line was purchased from the Pasteur Institute of Iran.
FGF7 Expression and Purification
Construction of codon optimized FGF7 plasmid
The codon optimization of FGF7 lacking the signal peptide sequence was obtained from Generay Biotech Co. Briefly, a 15% cutoff was used for codon efficiency, and any codon below 15% was removed except for positions with strong secondary structures checked using a build-in M-fold module. Internal ribosomal binding sites were removed. The codon-optimized gene was then verified for any interfering restriction sites with cloning followed by cloning in pET-28a (+) vector. Insertion of the FGF7’s gene was verified using gel electrophoresis via pET universal primers (pET 3′- CTAGTTATTGCTCAGCGG and pET 5′-TAATACGACTCACTATAGG).
Expression and purification of codon-optimized FGF7
The plasmid pET-28a (+) containing FGF7 was transformed into Escherichia coli BL21 (DE3) pLysS competent cells. LB media supplemented with kanamycin (50 µg/mL) was inoculated with the transformed bacteria and incubated overnight at 37 °C. The overnight culture was diluted in LB supplemented with kanamycin (50 µg/mL) at a ratio of 1:20 and further incubated at 37 °C with vigorous shaking until OD600 of 0.7. To the culture was added isopropyl β-D-1-thiogalactopyranoside (IPTG) at a concentration of 0.4 mM and incubated overnight at 25 °C at 150 rpm. Then, the culture was centrifuged at 5000 rpm for 10 min, and the harvested bacterial pellet was lysed using lysis buffer containing Tris 50 mM, NaCl 100 mM, β-mercaptoethanol 0.1%, lysosyme 0.1 mg/mL, and phenylmethane sulfonyl fluoride (PMSF) 1.4 mM. Subsequently, the lysate was frozen and thawed three times with further sonication on ice. The lysate was centrifuged at 10,000 rpm at 4 °C for 20 min. Then the supernatant was directly subjected to affinity chromatography using Ni-Sepharose beads. After several washing steps, FGF7 protein was eluted and dialyzed in phosphate-buffered saline (PBS) pH 7 buffer solution. Eluted FGF7 was applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for monitoring the expression and purification step.
Western blotting
Western blotting was carried out for detecting the expressed FGF7. Briefly, after resolving FGF7 by SDS-PAGE, the protein was transferred onto a polyvinylidene fluoride (PVDF) membrane at 250 mA for 90 min. The membrane was blocked with TBS-T (Tris 20 mM, NaCl 150 mM, Tween 0.05%, pH 7.5) buffer supplemented with 5% skim milk overnight at 4 °C. Then, the membrane was incubated with anti-His and goat anti-mouse HRP-conjugated antibodies as primary and secondary antibodies at 1:3000 and 1:9000 dilutions for 1.5 h, respectively. Several washing steps using TBS-T were performed between incubations. The bands corresponding to FGF7 were developed using the BM Chemiluminescence Western blotting kit.
Enzyme-linked immunosorbent assay–based FGF7 detection
Further verification of the expressed FGF7 was carried out by an enzyme-linked immunosorbent assay (ELISA) experiment. To this end, FGF7 protein was coated in MaxiSorb ELISA plates at the concentration of 100 µg/mL and incubated at 4 °C overnight. The next day, wells were washed three times with TBS and blocked with 5% bovine serum albumin (BSA) for 2 h at room temperature with gentle shaking. After the blocking step, the wells were washed three times with TBS and incubated with anti-FGF7 antibody as the primary antibody at a 1:500 dilution in 3% BSA for 1.5 h at room temperature under gentle shaking. After washing three times with TBS-T, the goat anti-mouse antibody at a 1:8000 dilution in 3% BSA was added and incubated for 1 h at room temperature. Finally, the substrate (TMB) was added, and the reaction was stopped by adding 1M sulfuric acid followed by recording the absorbance at 450 nm using a spectrophotometer.
Circular dichroism experiment
A sample of FGF7 (100 µg/mL) in 10 mM phosphate buffer at pH 7 was prepared for the circular dichroism (CD) spectropolarimetry experiment. The spectra were recorded on a CD215 (Aviv) spectropolarimeter operating with a temperature-controlled water bath. CD data were collected over the wavelength range of 190 to 260 nm, with a bandwidth of 1 nm. A quartz cuvette with an optical path length of 1 cm was employed, and spectra were recorded at 25 °C. The collected spectra were improved through background subtraction and then converted to mean residue molar ellipticity, [θ]MRE (deg cm2 dmol−1), based on equation (1):
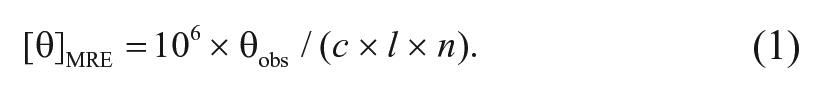
θobs is the observed ellipticity in mili degrees at the defined wavelength, c is the concentration of protein (µM), l is the optical length (mm), and n is the number of peptide bonds in the protein. Analyses of the secondary structure conformation based on the recorded CD spectra were performed using K2D, CDNN (version 2.1) and BeStSel. 27
In vitro scratch assay
To assess the healing property of FGF7, an in vitro scratch assay was performed. For this purpose, MCF-7 cells were cultured in RMPI supplemented with 10% FBS. The cells were seeded at 1.5 × 105 cells per well on 12-well plates and grown until 85% confluency at 37 °C in a 5% CO2 humidified atmosphere. The confluent cells were serum starved for 12 h. and then a cell-free area was created using a 200-µL pipette tip. After washing the cells with serum-free media, different concentrations of purified FGF7 (i.e., 100, 200 ng/mL) were added into the wells, and the incubation was continued for 24 h. The scratched area was photographed before and after treatment using an inverted phase-contrast microscope (Hund Wetzlar). ImageJ software (National Institutes of Health, Bethesda, MD) was used to quantify the restoration of the notched area.
Phage Display Procedure
Amplification and purification of phage antibody repertoire
The purchased human domain antibody library (dAb) 28 containing VH domains that were constructed in a pR2 phagemid with a diversity of 3 × 109 was diluted into 500 mL 2 × TY medium supplemented with 4% glucose and 100 mg/mL ampicillin and incubated at 250 rpm at 37 °C. After reaching an OD600 of 0.5, the amplified KM13 helper phage (1 × 1012 plaque-forming units [PFU]) was added to 250 mL culture and incubated at 37 °C for 60 min without shaking. Subsequently, the culture was centrifuged at 3200 g for 10 min and the pellet was resuspended in 500 mL 2 × TY medium supplemented with 0.1% glucose, 100 µg/mL ampicillin, and 50 µg/mL kanamycin and incubated overnight at 250 rpm at 25 °C. The overnight culture was centrifuged at 3200 g for 10 min, and the pellet was discarded. The phage in supernatant was isolated by the PEG precipitation method as described elsewhere. 28 The titer of the obtained phage was determined by infecting ER2738 bacteria with the phage at 37 °C using a 10-fold serial dilution method.
Panning procedure using the dAb phage library
Purified FGF7 protein with a 100 μg/mL concentration in TBS (50 mM Tris, 100 mM NaCl, pH 8.0) was coated in a 96-well plate. The plate was incubated at 4 °C overnight in an air-tight humidified box. Then, the wells were washed three times with TBS and blocked with blocking buffer (i.e., 2% skim milk in TBS) and incubated for 2 h at 4 °C. After incubation, the blocking buffer was removed by tapping the plate on tissue paper, and the wells were washed six times using TBS. A total amount of 5 × 1012 colony-forming unit (CFU) phages in TBS supplemented with 2% skim milk were added to the wells and incubated for 1 h at room temperature with gentle agitation. The supernatant was discarded, and the wells were washed 10 times with TBS containing 0.1% Tween-20 followed by an additional two washes with TBS. Then, 100 µL trypsin in TBS with a final concentration of 1 mg/mL was added to the wells and incubated at room temperature for 1 h by shaking on a rocking platform. The eluted phage particles were used for the next round of panning.
The eluted phage was added to a fresh culture of E. coli ER 2738 bacteria (OD600 of 0.4) and incubated for 30 min at 37 °C. After centrifugation of the culture at 3200 g for 5 min, the pellet was resuspended in 1 mL 2 × TY and plated equally on six LB plates supplemented with 100 µg/mL ampicillin and 4% glucose and incubated at 37 °C overnight. The titer of eluted phage was determined using the serial dilution method as mentioned before.
For the subsequent rounds of panning, cells infected with phagemid were scraped from agar plates by applying 5 mL 2 × TY medium using a glass spreader. The scraped cells mixed thoroughly by vortexing and diluted into 500 mL 2 × TY culture supplemented with 4% glucose and 100 µg/mL ampicillin followed by incubation at 37 °C at 250 rpm until reaching an OD600 of 0.5. A total of five rounds of panning were performed according to the protocol outlined above.
Sequence analysis of selected antibodies
From each round of panning, colonies grown on LB plates were randomly selected. Subsequently, 10 mL 2 × TY medium supplemented with 100 µg/mL ampicillin and 1% glucose was inoculated with single colonies and incubated overnight at 250 rpm at 37 °C. Colony PCR was performed using the primers (5′-CCCTCATAGTTAGCGTAACG-3′ and 5′-CAGGAAACAGCTATGAC-3′) for confirming the presence of domain antibody coding sequences in the samples, and the PCR products were sent out for sequencing.
Screening of clones by monoclonal phage ELISA
To estimate the binding affinity of the selected phage, an ELISA experiment was performed. To this end, FGF7 protein was coated in MaxiSorb ELISA plates with a final concentration of 100 µg/mL and incubated overnight at 4 °C. The next day, wells were washed three times with TBS and blocked with 3% BSA for 2 h at room temperature followed by washing three times with TBS. Then, the selected phage particles (which were produced in 96-well plates) were incubated with coated FGF7 for 1.5 h at room temperature with gentle shaking. After three washes with TBS-T, the HRP–anti-M13 antibody with a dilution of 1:5000 in 3% BSA was added and incubated for 1 h at room temperature. Then, TMB substrate was added, and the reaction was stopped by adding 1M sulfuric acid. Finally, the absorbance was measured at 450 nm by a microplate ELISA reader. To study the binding property of the phage harboring the domain antibody selected at the final round (i.e., dAb 5-10) to FGF7, the ELISA experiment was carried out at different concentrations of phage. Moreover, to further evaluate the affinity of the selected domain antibody toward FGF7, another ELISA experiment was conducted using different concentrations of coated FGF7 (i.e., 0.1, 0.3, 1, 3, 10, 30, and 100 µg/mL) against a constant concentration of phage dAb 5-10 (i.e., 2 × 109 CFU).
Results
Expression and Purification of Codon-Optimized FGF7
The purchased construct containing the codon-optimized FGF7 gene was verified by PCR reaction using pET universal primers. The PCR reaction result and the map of the construct are demonstrated in Supplemental Figure S1 . The vector harboring the codon-optimized FGF7 was transformed into E. coli BL21 (DE3) pLysS and expressed at the condition as outlined in the Materials and Methods section. The amount of purified FGF7 was 3.5 mg/L culture. The yield of purified protein is comparable to previous studies.29,30 Supplemental Figure S2A shows SDS-PAGE analysis of the protein after expression and purification. As can be seen in Supplemental Figure S2A , lane 1 corresponds to the Ni-Sepharose beads containing FGF7, whereas in lane 2, the purified eluted FGF7 with an expected size of 20 kDa is observed. Lane 3 shows the flow-through fraction with no observable FGF7 band.
Western Blotting
Further identification of FGF7 was conducted on purified protein using the western blot experiment. Since the recombinant FGF7 contained a 6x-His tag, an anti-His antibody was used for the immunoblotting. The result is shown in Supplemental Figure S2B , where a unique band of the protein is detectable at about ~20 kDa in all three lanes corresponding to the first and second elutions as well as Ni-Sepharose bead bound samples, respectively.
ELISA-Based FGF7 Detection
To investigate the binding ability of the purified FGF7, an ELISA experiment was carried out. As shown in Supplemental Figure S3 , the purified 6x-His–tagged FGF7 is detectable specifically by both anti-FGF7 and anti–6x-His antibodies, respectively, while the negative controls did not lead to an appreciable signal.
Folding Assessment of FGF7
The verification of proper folding of the codon-optimized FGF7 was assessed by the CD experiment. The sample for the CD experiment was prepared freshly by dialyzing the affinity-purified protein against PBS (pH 7.4). Supplemental Figure S4 and Supplemental Table S1 show the results of the CD experiment, which are in agreement with the secondary structure content of FGF7 previously reported by the CD experiment4,31 or X-ray crystallography. 32 The CD spectrum of the purified FGF7 compares very well with that obtained for correctly folded FGF7 reported by Buchtova et al. 31 and shows substantial differences from its denatured form (see Figure 5A in Buchtova et al. 31 ).
In Vitro Cell Migration Assay
The scratch assay was performed to evaluate the healing functionality of produced FGF7. To do this, the confluent cells were serum starved to retard the growth rate and hence make it possible to investigate the proliferative effect of FGF7. After introducing a gap by scratching the monolayered cells, the treatments were applied and the cells were incubated overnight. The migration of the cells was photographed ( Suppl. Fig. S5 ) and quantified using ImageJ software. The percentage of migration (monolayer wound-healing effect) significantly (p < 0.05) increased by adding either 100 or 200 ng/mL FGF7 compared to the untreated controls ( Suppl. Fig. S6 ). However, the extent of cell migration at the used concentrations of FGF7 was not statistically different.
Phage Display Library Panning
Phage display technique was employed to identify novel single-domain antibodies against FGF7 in five consecutive rounds of panning. Phage recovery at each round of selection was determined by titrating eluted phage before amplification as outlined above. As shown in Table 1 , the titer of the phage particles increased after the first round of panning and reached an almost constant titer. Increasing the phage titer during the panning process is indicative of enrichment in the selected dAbs.
Panning Efficacy of Domain Antibody Library against FGF7.
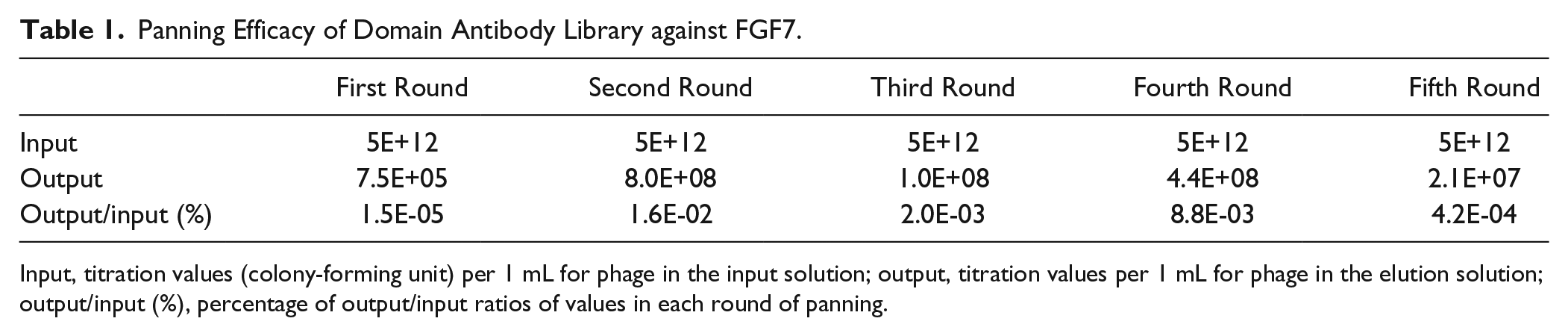
Input, titration values (colony-forming unit) per 1 mL for phage in the input solution; output, titration values per 1 mL for phage in the elution solution; output/input (%), percentage of output/input ratios of values in each round of panning.
Sequence Analysis of the Selected Domain Antibodies
The isolated colonies from each round of screening were randomly selected, and after confirming the presence of the sequence coding for the domain antibody with the expected size (~650 bp) using colony PCR, the products were sequenced. The sequences of the identified domain antibodies were aligned using the Clustal Omega program. Complementarity-determining regions (CDRs) were inspected in domain antibodies and analyzed for any pattern of enrichment or similar physicochemical properties ( Table 2 ). As can be seen in Table 2 , CDR3 is the most variable region of the selected clones with numerous indels (insertions and deletions), while CDR2 is the least variable region.
Complementarity-Determining Regions (CDRs) of Selected Phage Particles.
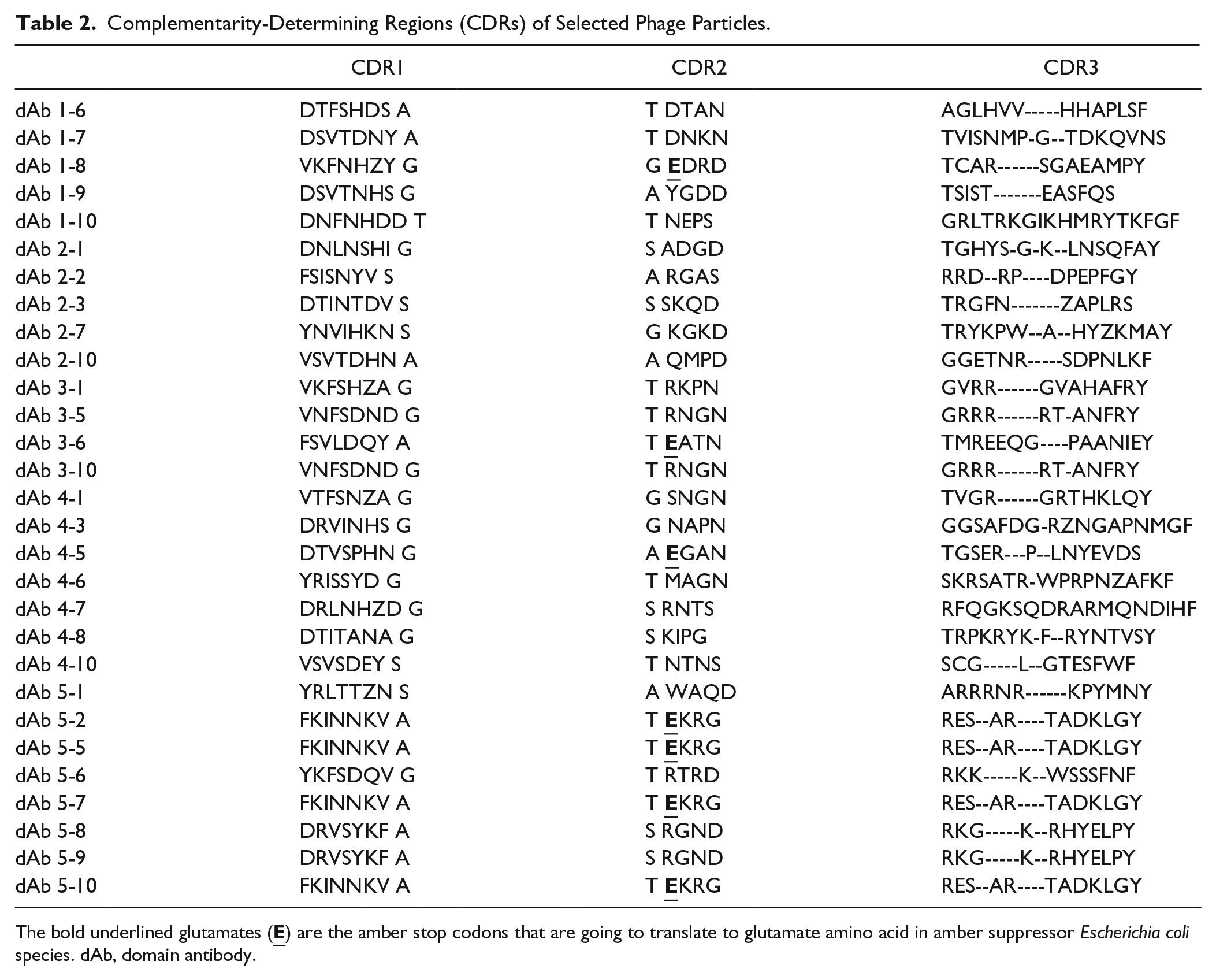
The bold underlined glutamates (
Isolation of Specific FGF7 Binding Phage Clones
After five rounds of panning, the phage particles with affinity toward FGF7 were recovered and screened by an ELISA experiment to identify the domain antibodies specific to FGF7. As shown in Figure 1 , nine different domain antibodies were selected from the three last rounds of panning and used for the ELISA experiment. To assess the concentration-dependence of FGF7 binding, one of the selected phage particles (dAb 5-10) was subjected to an ELISA experiment at four different concentrations, and the result is shown in Figure 2 . The rational for selecting dAb 5-10 for further investigation was its intensity in the ELISA experiment and relatively more frequent appearance at the fifth round of panning. In addition, the affinity of dAb 5-10 toward FGF7 was evaluated by preparing FGF7-coated ELISA plates using FGF7 solutions with different concentrations ranging from 0.1 to 100 µg/mL. The results demonstrated that phage displaying dAb 5-10 is capable of detecting FGF7 in wells that were coated by FGF7 solution with a minimum concentration of 3 µg/mL ( Figure 3 ).
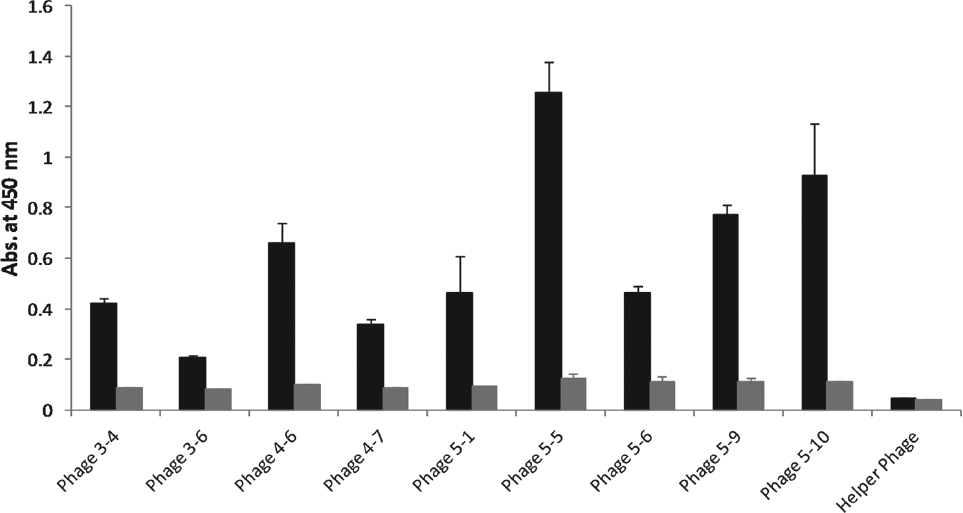
Evaluation of fibroblast growth factor 7 (FGF7) binding capability of a selected single-domain antibody displaying phage particles using the enzyme-linked immunosorbent assay (ELISA) experiment. Briefly, FGF7 protein was coated in MaxiSorb ELISA plates with a final concentration of 100 µg/mL. The coated wells were incubated with the selected phage particles. Then, the bound phage particles were detected by horseradish peroxidase (HRP)–anti-M13 antibody. Helper phage was used as a negative control for the experiment. The black columns are the results for wells with coated FGF7 while the gray columns are blanks where the wells were not coated by FGF7. The absorbance values for the coated wells are significantly (p < 0.05) higher than that for the uncoated wells, except for the negative control.
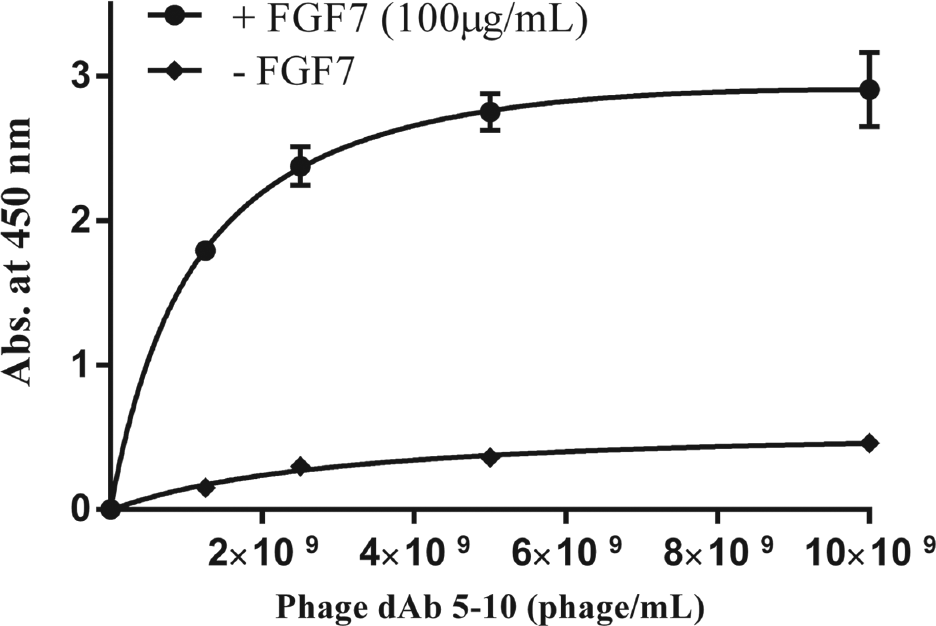
Evaluation of concentration dependency of fibroblast growth factor 7 (FGF7) binding capability of dAb 5-10 single-domain antibody displaying phage using the enzyme-linked immunosorbent assay (ELISA) experiment. The ELISA experiment was conducted for different concentrations of the dAb 5-10 phage particle against a constant concentration of coated FGF7 (i.e., 100 µg/mL) as described in the Materials and Methods section.
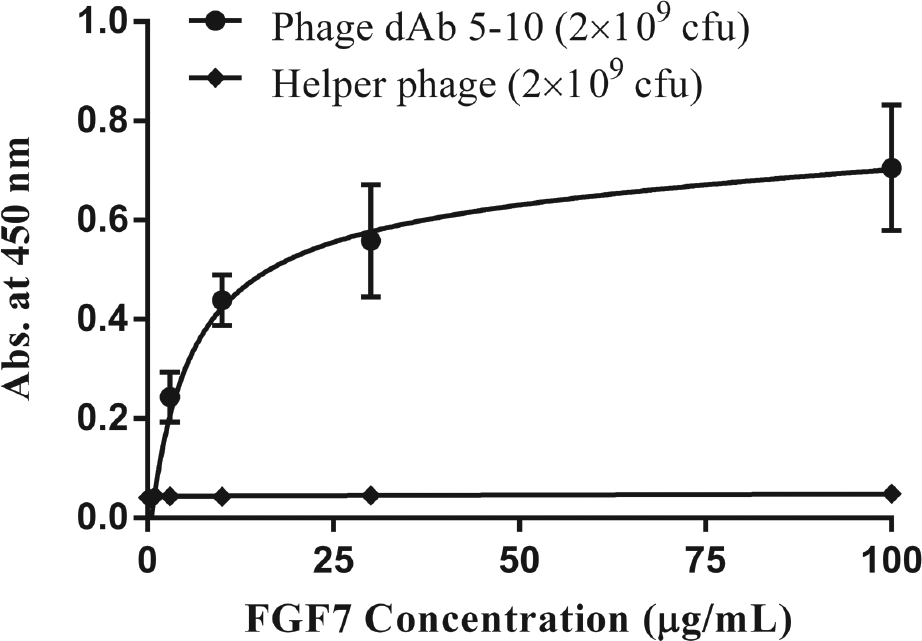
Evaluation of dAb 5-10 affinity to different concentrations of fibroblast growth factor 7 (FGF7). The enzyme-linked immunosorbent assay (ELISA) experiment was conducted using different concentrations of coated FGF7 (i.e., 0.1, 0.3, 1, 3, 10, 30, and 100 µg/mL) against a constant concentration of phage dAb 5-10 (i.e., 2 × 109 colony-forming units) as described in the Materials and Methods section. Helper phage was used as control for the experiment.
Discussion
FGF7 belongs to fibroblast growth factor family with potent proliferative and differentiating activity. FGF7 binds selectively to fibroblast growth factor receptor 2 isoform IIIb with a relative specificity of 80%. 33 The only Food and Drug Administration (FDA)–approved form of fibroblast growth factors on the market is palifermin, which is used for the treatment of oral mucositis induced by chemotherapy and radiotherapy procedures. 2 However, the use of palifermin in solid tumors is not recommended because of FGF7 receptor (FGFR2b) overexpression in epithelial tumor cells. 5 Moreover, CAFs and their associated components along with tumor cells have been proposed as therapeutic targets in cancer therapy. 34 Several lines of evidence show that FGF7, one of the vital factors of CAFs, contributes significantly to vasculation and angiogenesis in tumors. Therefore, inhibition of FGFs and their corresponding receptor (FGFR 2IIIb) provides remarkable effects in reducing angiogenesis, cell proliferation, and cancer progression. 35
The codon-optimized FGF7 was expressed in E. coli BL21 (DE3) pLysS and purified by Ni-Sepharose affinity chromatography as outlined above. The purity of the produced FGF7 was approximately 92%. The yield of codon-optimized His-FGF7 was 3.5 mg/L of culture, which is close to the yields reported for FGF7-His (0.6 mg/L) and FGF-HaloTag (5.4 mg/L) proteins. 30 Western blot and ELISA experiments were used to characterize the purified FGF7. The results shown in Supplemental Figure S2B and Supplemental Figure S3 indicate that the produced FGF7 can be used as an appropriate matrix during the panning procedure.
CD spectroscopy was the other technique used to study the folding of the produced FGF7. The calculated secondary structure content for the produced FGF7 in the solution was in close agreement to that assigned based on its X-ray crystal structure (PDB code: 1QQK) analyzed using DSSP and STRIDE algorithms, as well as the results of CD experiments by others.4,31
Phage display as a high-throughput technique has earned considerable interest for the identification of novel antibodies against various targets. There are few reports where this technique was used to identify new peptides against members of the FGF family of proteins. For instance, Yayon et al. 36 identified a new peptide that inhibits the binding of basic fibroblast growth factor (bFGF) as an important protein involved in the progression and angiogenesis in solid tumors to its receptor. Following inhibition, the proliferation of endothelial cells was suppressed. In another study conducted by Wu et al., 37 a novel basic FGF-binding peptide with potent antiangiogenic activity was discovered. In this study, the isolated P7 peptide (PLLQATL) was capable of binding to bFGF through interrupting the interaction of the protein to its receptor. Similarly, bFGF-mediated proliferation and angiogenesis were inhibited. In another study, for the purpose of specific delivery of gene therapy vectors, heptapeptides with the consensus motif of MXXP were identified with a high affinity to the FGF receptor. 38 Maruta et al. 38 showed that the MQLPLAT peptide caused a 40-fold increase in transgene transduction of polyelectrolyte gene delivery. Fan et al. 39 used FGFR1 as a potential target for cancer therapy and isolated peptides showing significant inhibition of mitogenic activity of FGF by screening a 6-mer phage display peptide library against Sf9 insect cells expressing FGFR1.
In the current work, the selection of single-domain antibodies against FGF7 was sought using the phage display approach. For this purpose, we have used the dAb library for panning against the purified FGF7. After five rounds of panning, several colonies were randomly selected and sent out for sequencing. CDR1, which encompasses eight variable residues, showed diversity in sequence; however, at some positions, amino acids with similar physicochemical properties were present. For example, in some clones, aromatic amino acids (i.e., Phe/Tyr) were observed at position 1. At position 2, basic amino acids (i.e., Lys/Arg) could be seen, especially for those isolated at the final rounds of selection (i.e., 4 and 5), followed by hydrophobic residues (i.e., Val/Ile/Phe) at position 3. At position 4 of the CDR1, nucleophilic residues (i.e., Ser/Thr) were prevalent, but threonine was substituted with asparagine for those clones isolated at the final rounds of panning. The first position of CDR2 mostly held the stop codons, which were translated to glutamine in amber suppressor E. coli strains such as the one used in this work. Arginine and asparagine at position 3 of CDR2 and some small residues (i.e., Asp/Asn/Gly) at position 4 of CDR2 were conserved. CDR3 was the most variable region, with many indels (insertions and deletions) leading to its variable length among different phages, yet some hydrophobic residues were detectable at positions 5, 7, 9, and 11.
In the next step, phage from the three last rounds of panning were subjected to the ELISA experiment. The results showed that dAb 5-5, 5-9, and 5-10 had a relatively high intensity in the ELISA experiment, which may indicate their stronger binding to FGF7 relative to the other selected clones. However, one should bear in mind that the higher ELISA intensities do not necessarily translate to stronger affinities due to the differences in phage yields obtained from the different clones of sdAb. Among these three clones, dAb 5-10 was more frequent than the others and hence was selected for further investigation. To determine the binding characteristics of dAb 5-10, the concentration-dependent ELISA experiment was performed ( Fig. 2 ), indicating the effective binding between dAb 5-10 and FGF7.
The vasculation and angiogenesis effects of FGF7 in solid tumor development have been proved.11,13,34,40 Therefore the development of anti-FGF7 agents may find application in cancer therapy. According to the literature, this is the first time that FGF7, a proliferative agent in tumor progression, was targeted using phage displayed antibodies. The newly identified single-domain antibodies may serve as promising tools for developing tumor-suppressing agents via inhibiting the FGF7 tumorigenic effects.
Footnotes
Acknowledgements
The authors thank the respectful reviewers for their valuable comments and suggestions.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We thank the Research Office and Biotechnology Research Center of Tabriz University of Medical Sciences for providing financial support under the Postgraduate Research Grant scheme for the PhD thesis by Behzad Jafari.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.