
Abstract
Although diverse efforts have been done to identify biomarkers for control of tuberculosis using laboratory strain Mycobacterium tuberculosis H37Rv, the disease still poses a threat to mankind. There are many emerging M. tuberculosis strains, and proteomic profiling of these strains might be important to find out potential targets for diagnosis and/or prevention of tuberculosis. We evaluated the comparative proteomic profiling of culture filtrate (CF) proteins from prevalent M. tuberculosis strains (Central Asian or Delhi type; CAS1_Del, East African-Indian; EAI-3 and Beijing family) by 2D polyacrylamide gel electrophoresis and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry. As a result, we could identify 12 CF proteins (Rv0066c, Rv1310, Rv3375, Rv1415, Rv0567, Rv1886c, Rv3803c, Rv3804c, Rv2031c, Rv1038c, Rv2809, and Rv1911c), which were consistently increased in all prevalent M. tuberculosis strains, and interestingly, two CF proteins (Rv2809, Rv1911c) were identified with unknown functions. Consistent increased intensity of these proteins suggests their critical role for survival of prevalent M. tuberculosis isolates, and some of these proteins may also have potential as diagnostic and vaccine candidates for tuberculosis, which needs to be further explored by immunological analysis.
Introduction
The global burden of tuberculosis (TB) disease (caused by Mycobacterium tuberculosis) still continues, and according to the World Health Organization (WHO), 10.4 million new TB cases and 1.8 million deaths have been estimated annually. 1 Various attempts have been done to find out potential markers against TB using H37Rv and Erdman strains by proteomic approaches,2–4 but there are few reports about proteomic profiling of emerging clinical strains of M. tuberculosis.5,6 This issue is of particular importance as in vitro culture passaging of the laboratory strain H37Rv can reduce its virulent factors. 7 It has also been shown that the proteome of M. tuberculosis H37Rv may differ with response to environmental conditions as well as host interaction.8,9 Therefore, proteomic analysis of the M. tuberculosis strains isolated from the population of a particular region may help in identifying some proteins that could be useful as a diagnostic or vaccine candidate for TB. In addition, the sensitivity of available molecules for early specific diagnosis and the potential for protection against disease are compromised due to wide variation of immune responses to M. tuberculosis in humans. Our previous study showed that clinical isolates belonging to prevalent genotypes (Central Asian family or Delhi type; CAS1_Del, East African Indian family; EAI-3 and Beijing family) induced better antibody and lymphoproliferation as well as cytokine secretion in peripheral blood mononuclear cells from PPD+ healthy controls compared to H37Rv. 10 The study has been further extended to identify dominant culture filtrate protein(s) from these prevalent clinical isolates of M. tuberculosis in north India. These proteins might be used as future candidates for diagnostics, vaccine, and drug targets for TB.
Materials and Methods
Mycobacterium tuberculosis Isolates
M. tuberculosis isolates and H37Rv were obtained from the Mycobacterial Repository Centre, National Reference Laboratory, National JALMA Institute for Leprosy & Other Mycobacterial Diseases (ICMR), Tajganj, Agra, India. M. tuberculosis strains were genotyped by IS6110 DNA fingerprinting and spoligotyped as described previously.11,12 M. tuberculosis isolates belonging to prevalent genotypes (Central Asian family or Delhi type; CAS1_Del, East African Indian family; EAI-3 and Beijing family) and showing strong immune response were selected from our previous study. 10 M. tuberculosis isolates were inoculated (approximately 108 colony-forming units [CFU]/mL) in Tween-80 free Sauton’s medium (Himedia, India) and incubated at 37 °C for 3 weeks. All isolates were grown in biological replicates.
Preparation of Culture Filtrate Proteins
Culture filtrate proteins were prepared as described earlier.13,14 In brief, culture was centrifuged at 18,000 × g for 30 min at 4 °C and sequentially filtered through a 0.45-µm followed by a 0.22-µm Millex GV PVDF membrane (Millipore, Bedford, MA). Then, 10% sodium dodecyl sulfate (SDS) (Sigma, St. Louis, MO) was added to obtain 0.1% final concentration (w/v) in culture filtrate (CF) and kept in a boiling water bath for 5 min. CF was treated with trichloroacetic acid (TCA, w/v) (Sigma) to get a final concentration of 10% (w/v). Finally, this mixture was incubated at −20 °C for 5 h, and resulting precipitate was removed by centrifugation at 18,000 × g for 30 min at 4 °C. Minimum volume of high-performance liquid chromatography (HPLC) grade water (Qualigens Fine Chemicals, Mumbai, India) was added to disperse the pellet, and then whole suspension was washed with 1 mL prechilled acetone (Sigma-Aldrich, St Louis, MO). Air-dried pellets were dissolved in a minimum volume of 2D sample preparation buffer (Bio-Rad Laboratories, Hercules, CA). These extracts were stored at −20 °C until used. Protein concentration was determined by Bradford’s method. 15 Protein extractions and quantifications were done in technical replicas.
2D Polyacrylamide Gel Electrophoresis
Isoelectric focusing (IEF) was done using the method of Gorg et al. 16 and Sharma and Bisht. 17 In brief, 100 µg CF protein in 2D sample preparation buffer (125 µL) was pipetted into each well of the rehydration tray. IPG strips were allowed to rehydrate for 16 to 18 h at 20 °C. IEF run was performed on the IEF cell (BIO-RAD, Hercules, CA, USA) using (1) linear mode, 0 to 250 V, 1 h; (2) rapid mode, 250 V, 1.5 h; (3) linear mode, 250 to 3000 V, 4 h; (4) rapid mode, 3000 V constant until 15 kVhr; and (5) slow mode, 500 V, 10 h. Temperature was set at 20 °C, and the current limit was 50 µA/IPG strip. After completion of the first dimension run, each strip was equilibrated for 10 min with equilibration buffer I and equilibration buffer II (BIO-RAD, Hercules, CA, USA). IPG strips were placed onto the surface of 12% SDS polyacrylamide gel and sealed with 1% agarose in electrode buffer containing bromophenol blue. Gel was run at a constant 100 V until the dye front just migrated off the bottom of the gel. Gel was then stained with 0.2% Coomassie brilliant blue R-250 (Sigma-Aldrich, St Louis, MO) dye for 3 h and destained to visualize the protein spots. 2D gel was run in technical replicates and analyzed by the Gel Documentation system (BIO-RAD, Hercules, CA, USA) using PDQuest Software (BIO-RAD, Hercules, CA, USA).
In-Gel Digestion
In-gel digestion of proteins spots with trypsin was performed using the protocol by Shevchenko et al. 18 In brief, protein spots were excised using a Propic spot picker (Genomic Solutions Ltd., Huntingdon, UK) and digested by a protein digester (Genomic Solutions Ltd.). Excised gel slices were washed with HPLC-grade water (Merck, Darmstadt, Germany) for 10 min followed by destaining with 50% ACN (Loba Chem, Mumbai, India) in 25 mM ammonium bicarbonate (Sigma). The gel slices were dehydrated by incubating them with 50 µL of 100% ACN for 10 min at room temperature (RT). Dried slices were reduced using 10 mM dithiothreitol (DTT) (Sigma) at 56 °C for 45 min and alkylated with 55 mM iodoacetamide (Sigma), both in 100 mM ammonium bicarbonate. The gel slices were again dehydrated by 100% ACN as described above and rehydrated in 25 mM ammonium bicarbonate containing modified trypsin (200 µg/protein spot) (Promega, Madison, WI) at 4 °C for 30 min on ice. Ammonium bicarbonate (25 mM) was added to gel slices and incubated at 37 °C for 16 to 20 h in a humid chamber. Resulting peptides were collected in a fresh tube, and gel slices were sonicated with 1% TFA in 100% ACN solution (1:1) in a water bath sonicator. The volume of peptide solution was reduced in the vacuum centrifuge CentriVap Concentrator (LABCONCO, Kansas City, MO). Peptides were eluted on a matrix-assisted laser desorption ionization (MALDI) target plate with α-cyano hydroxyl cinnamic acid (Sigma) through Zip Tips (Millipore, Billerica, MA) using the manufacturer’s protocol.
MALDI–Time-of-Flight Analysis
Mass spectra of digested proteins were acquired using an Autoflex II TOF/TOF 50 (Bruker Daltonik GmbH, Leipzig, Germany) in positive reflectron mode. Instrument was made ready with a 337-nm nitrogen laser and a 50-Hz digitizer, and percentage of laser energy was kept at 30% to 40%. Pulse energy was 105 µJ with a 1.3-ns pulse duration. Final mass spectra were produced by averaging 2000 to 3500 laser shots taken at different positions within each spot. Spectra were acquired in positive reflectron mode in the detection mass range of 500 to 3000 m/z. The machine was calibrated by using Peptide Calibration Standard II (Bruker Daltonics GmbH). MALDI–time-of-flight mass spectrometry (MALDI-TOF/MS) spectra were processed and analyzed with flexAnalysis 2.4 software (Bruker Daltonics GmbH). Initially, spectra acquired were processed for baseline subtraction with 80% baseline flatness followed by smoothening with threshold of signal-to-noise ratio (S/N) >5. The contaminant m/z peaks originating from human keratin, trypsin auto-digestion, and matrix were removed from the spectra to generate the peptide mass list for the database search. Acquired mass spectra were processed through the flexAnalysis 2.4 program for peak detection and submitted to the Mascot server using the Mascot Wizard program (Matrix Science, London, UK) for identification. Proteins were searched against the available M. tuberculosis complex database in Swissprot using the MASCOT program. Peptide mass tolerance was set in the range of 50 to 100 ppm (to acquire the best MASCOT score) with carbamidomethyl-cystein set as fixed modification, oxidation of methionine as variable modification, and 0 or 1 missed cleavage site allowed.
Signal Peptide Prediction
Signal peptides were predicted by the neural network (NN) and the hidden Markov model (HMM) methods using the SignalP 4.0 server (www.cbs.dtu.dk/services/SignalP).
Protein-Protein Interaction
The STRING-10 server was used to predict the interacting partners of protein-protein interaction.19–21 STRING-10 uses a combination of prediction approaches and an integration of other information (neighborhood, transferred neighborhood, gene fusion, co-occurrence, coexpression, experiments, databases, text mining). Network was made at a medium confidence level (0.400), allowing all active prediction methods.
Results
Our previous published study showed that prevalent genotypes of M. tuberculosis clinical isolates (CAS Del, East African Indian and Beijing family) induce better antibody and T-cell responses compared to H37Rv. 10 CF proteins of these clinical isolates and H37Rv were compared by 2DE (2-dimensional gel electrophoresis). The composite 2D gel images of CF proteins are shown in Figure 1 . Protein spots from Coomassie brilliant blue–stained 2D polyacrylamide gel electrophoresis (2D-PAGE) gel were excised and subjected to in-gel trypsin digestion followed by MALDI-TOF/MS analysis. Selection of spots was based on their consistent increased intensities in all prevalent clinical isolates (cutoff limit ≥1.5-fold change in spot intensity). Student t test was used for the statistical analysis by PDQuest Advanced software (Bio-Rad, Hercules, CA). The system picks up the spots with differential intensity of significant levels built in the system. 17 The reproducibility of 2D-PAGE for CF preparations was confirmed by more than three different batches of collected CF of a clinical isolate. Protein spots of increased intensities were identified by MALDI-TOF/MS ( Table 1 ). A representative mass spectrum of the protein spot is shown in Figure 2 . The identified proteins were classified according to the M. tuberculosis H37Rv gene annotation database (http://genolist.pasteur.fr/TubercuList/) and belonged to five functional groups. The protein with highly acidic nature was probable adenosine triphosphate (ATP) synthase protein: Rv1310 (spot 3, pI 4.8) as observed by 2D-PAGE/MALDI-TOF/MS. Proteins with the lowest molecular mass (10.99 kDa) and highest molecular mass (82.84 kDa) were identified as excretory secretory/esx J (spot 11) and probable isocitrate dehydrogenase/icd2 (spot 1) respectively.
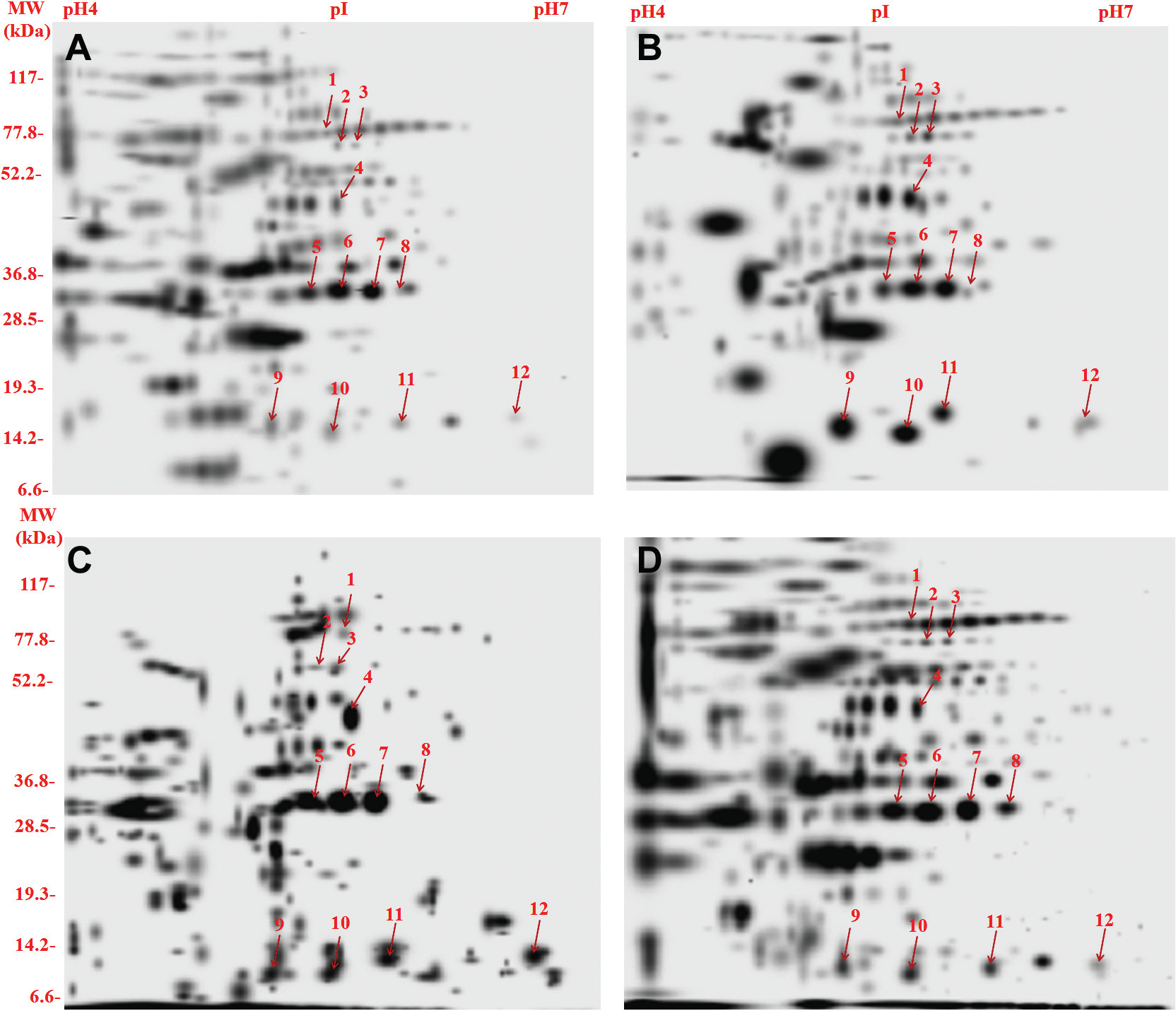
2D polyacrylamide gel electrophoresis of culture filtrate proteins (100 µg/gel) of prevalent families of Mycobacterium tuberculosis clinical isolates and H37Rv. (
Details of Culture Filtrate Proteins of Mycobacterium tuberculosis Showing Increased Intensities and Identified by MALDI-TOF/MS and MS/MS.
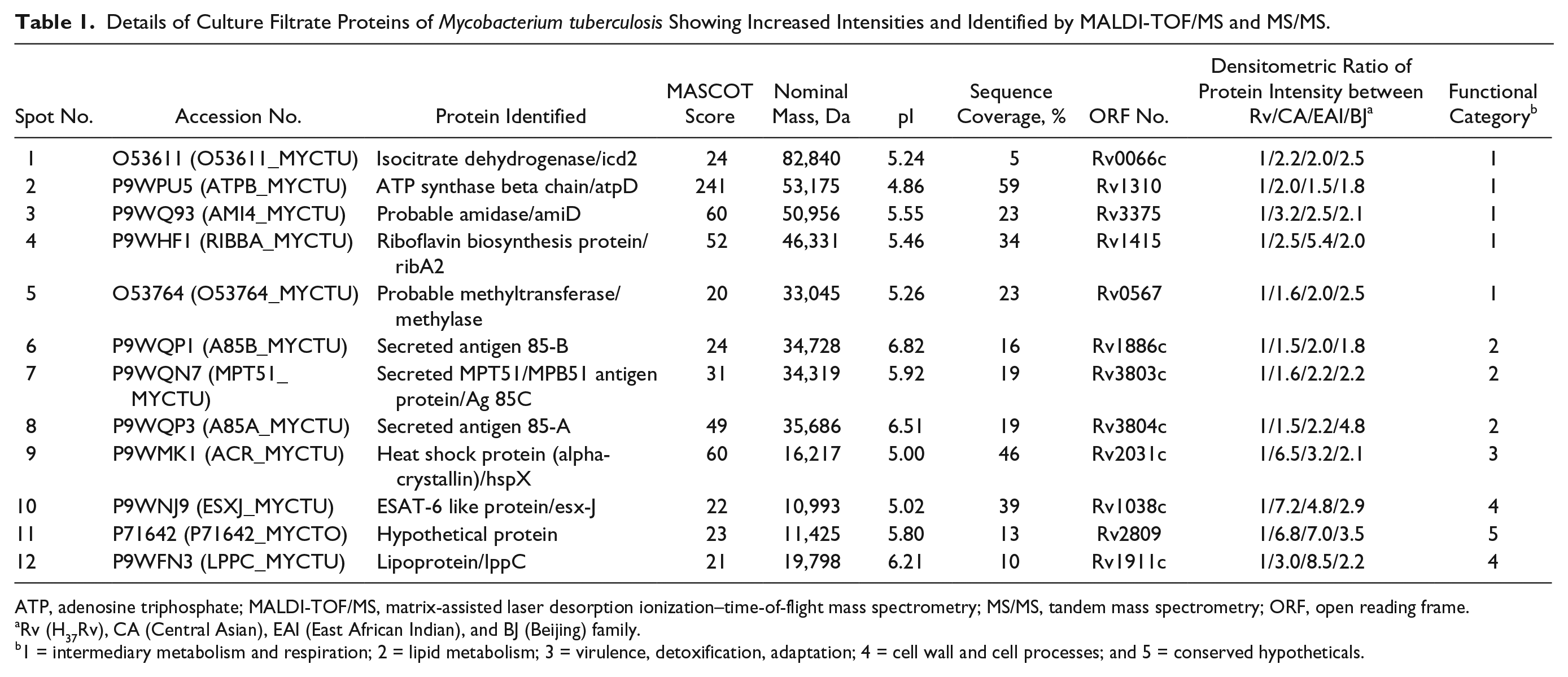
ATP, adenosine triphosphate; MALDI-TOF/MS, matrix-assisted laser desorption ionization–time-of-flight mass spectrometry; MS/MS, tandem mass spectrometry; ORF, open reading frame.
Rv (H37Rv), CA (Central Asian), EAI (East African Indian), and BJ (Beijing) family.
1 = intermediary metabolism and respiration; 2 = lipid metabolism; 3 = virulence, detoxification, adaptation; 4 = cell wall and cell processes; and 5 = conserved hypotheticals.
A representative mass spectrum of the protein spot.
We have identified 12 proteins, with consistently increased intensities in all three prevalent clinical isolates of M. tuberculosis. These were isocitrate dehydrogenase (icd2; Rv0066c), ATP synthase beta chain (atpD; Rv1310), amidase (amiD; Rv3375), riboflavin biosynthesis protein (ribA2; Rv1415), probable methyltransferase (Rv0567), Ag 85B (Rv1886c), MPT51/Ag 85C (Rv3803c), Ag 85A (Rv3804c), heat shock protein/hspX (alpha-crystallin; Rv2031c), ESAT-6–like protein (esx-J; Rv1038c), hypothetical protein (Rv2809), and probable lipoprotein (lppC; Rv1911c), and they are shown in Table 1 . Among 12 identified proteins, five are involved in intermediary metabolism and respiration, three in lipid metabolism, and two in cell wall and cell processes, one in virulence, detoxification, adaptation, and one as a hypothetical protein ( Table 1 ). Of 12 identified proteins, only four had signal peptide predicted (lppC, Ag85A, Ag85B, and Ag85C) using one or both of the NN and HMM methods (www.cbs.dtu.dk/services/SignalP). STRING analysis predicted ( Fig. 3 ) that proteins involved in intermediary metabolism and respiration, lipid metabolism and virulence, and detoxification and adaptation category not only interacted with each other but also to their protein partners.
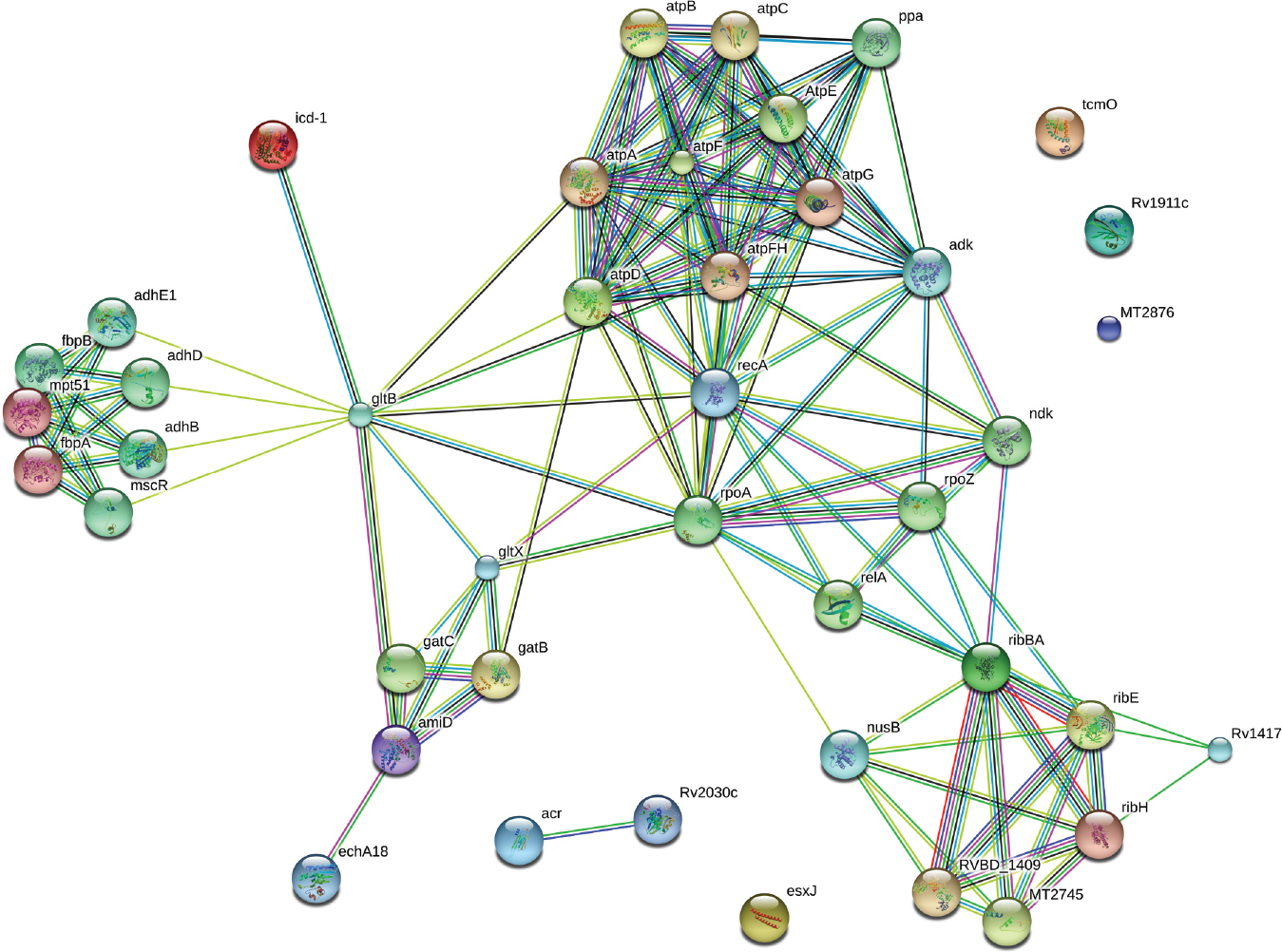
STRING-10 (an online database) was used for interactome analysis, which revealed the interacting partners of the overexpressed proteins, which hold key pathways involved in the pathogenesis of the prevalent strains of Mycobacterium tuberculosis.
Discussion
Comparative proteomic profiling of prevalent M. tuberculosis strains may be promising to determine the consistent potential markers for TB diagnosis, protection, and pathogenesis. Proteins secreted by M. tuberculosis are believed to be immunologically important as these molecules interact early with host cells and are therefore potentially important for virulence and pathogenesis of TB. 22 Comprehensive proteome profiling of M. tuberculosis using CF proteins is a tedious task because the low abundance of these proteins in media makes their recovery difficult, and various compounds in media further complicate their purification. Therefore, studies using CF proteins of M. tuberculosis are still limited. Mattow et al. 23 have done comprehensive proteome analysis and reported six unique CF proteins in M. tuberculosis H37Rv using high-resolution 2DE followed by MS and N-terminal sequencing. To date, little attention has been given to compare protein profiles of recent clinical M. tuberculosis isolates and laboratory strain H37Rv.24,25 Variable phenotypic differences 26 have been reported for a variety of M. tuberculosis strains that could be responsible for the outcome of M. tuberculosis infection. 27 These findings suggest that some strains of M. tuberculosis are more virulent, and the reason for this variability between strains may be their different antigenic repertoire.
Proteomic analysis in our study revealed consistently increased intensity of 12 proteins in prevalent clinical isolates (CAS1_Del, EAI and Beijing) in comparison to laboratory strain H37Rv. These were identified as Icd2/Rv0066c, atpD/Rv1310, amiD/Rv3375, ribA2/Rv1415, Rv0567, Ag 85B/Rv1886c, MPT51/Ag85C/Rv3803c, Ag 85A/Rv3804c, hspX/Rv2031c, esx-J/Rv1038c, Rv2809, and lppC/Rv1911c.
Rv0066c (isocitrate dehydrogenase [ICD]) is a key regulatory enzyme in the Krebs cycle. The immunodominant nature of ICDs is noted to discriminate extrapulmonary cases from healthy subjects. A study regarding the specificity of ICDs to differentiate TB patients from BCG-vaccinated cases and persons infected with mycobacterium other than TB (MOTT) has been reported. 28 Recently, Guirado et al. 29 reported overexpression of the ICD gene during latent TB. Rv1310/ATP synthase enzyme is responsible for ATP production by oxidative phosphorylation and therefore essential for survival of a microorganism. 30 Drug discovery trials for TB treatment are under way by targeting this enzyme. This protein was noted to be upregulated in M. tuberculosis–infected macrophages. 31 Recently, Sharma et al. 32 reported increased intensity of ATP synthase in aminoglycoside-resistant M. tuberculosis clinical isolates. Spot 3 was identified as Rv3375/probable amidase. amiD gene has been shown to be essential during infection of mice. 33 Rv1415/probable riboflavin biosynthesis protein (ribA2) was increased in our study and is likely to be involved in riboflavin metabolism. Singhal et al. 34 reported an increased intensity of this protein in Mycobacterium bovis BCG-infected macrophages. Spot 5 was identified as probable methyltransferase (Rv0567). To our knowledge, there is no information available for this protein.
Furthermore, increased intensity of secreted Ag85A (3804c), Ag85B (Rv1886c), and Ag85C (Rv3803c) was noted in prevalent clinical isolates. Similar to our finding, increased intensity of these proteins was also reported in prevalent Beijing strains.5,24 Recently, Sharma and Bisht 35 reported the overexpression of Rv3804c in the secretory proteome of streptomycin-resistant clinical isolates. Alpha-crystallin/hspX (Rv2031c), a heat shock protein, is involved in cell protection to various stimuli, such as stress, dormancy, heat, drugs, and hypoxia, by preventing protein aggregation. Pheiffer et al. 24 have reported increased expression of hspX in the Beijing strain. Shin et al. 25 have noted increased expression of hspX in strain K, which is dominant in Korea. Recently, Sharma et al. 32 reported increased intensity of Rv2031c in aminoglycoside-resistant M. tuberculosis clinical isolates. Rv1038c, an ESAT-6–like protein/esx-J, was increased in the Beijing strain. 5 Comparative proteomic analysis of CF proteins from M. tuberculosis K and other virulent strains, CDC1551 demonstrate that ESAT-6 was observed more often in virulent strains as compared with H37Rv. 36 Rv1911c, a probable lipoprotein (lppC), represents a class of proteins abundantly found in the cell membrane of mycobacteria and is known for virulence of M. tuberculosis. 37 Intensity of Rv2809 (spot 11) was also consistently increased in all isolates and identified as a conserved hypothetical protein with unknown function. Recently, Sharma and Bisht 38 emphasized that conserved hypothetical proteins might be involved in virulence/pathogenesis/resistance and could be used as future diagnostics or drug targets. Additional efforts are needed to assess its role in M. tuberculosis infection.
We hypothesized that Rv0066c, Rv1310, Rv3375, Rv1415, Rv0567, Rv1886c, Rv3803c, Rv3804c, Rv2031c, Rv1038c, Rv2809, and Rv1911c might be involved in virulence/pathogenesis of prevalent strains and could have potential as drug targets/diagnostics/vaccine candidates against TB, which needs to be evaluated.
Among the 12 identified proteins, four proteins showed the signal sequence using the NN or HMM method by SignalP 4.0 server (www.cbs.dtu.dk/services/SignalP/). Malen et al. 39 showed that most of the identified N-terminal peptides of mature secreted proteins had an AXA motif N-terminally to their cleavage sites and suggested that the mycobacterial signal peptidase I preferentially recognizes the AXA motif, which is underestimated by the SignalP training set. This could be an explanation for failure to identify the signal peptide of the other CF proteins in our study. Moreover, another study showed that several CF proteins seem to be exported by a sec-independent pathway, such as ESAT-6, superoxide dismutase, and glutamine synthase. 40
STRING analysis revealed that overexpressed proteins of intermediary metabolism and respiration, lipid metabolism, virulence, detoxification, adaptation, cell wall and cell processes, and conserved hypotheticals interacted with the other proteins, which were involved in various pathways of these categories. 20 We suggested that overexpressed proteins along with their interactome hold key pathways, which might cumulatively be involved in the pathogenesis of the prevalent strains of M. tuberculosis. In conclusion, comparative proteomic profiling of CF proteins of prevalent clinical M. tuberculosis isolates of north India by 2D-PAGE and MALDI-TOF/MS revealed consistently increased intensities of 12 proteins in all prevalent isolates, and of these, two are annotated with an unknown function. These CF proteins might have a role in survival/pathogenesis of prevalent strains of M. tuberculosis, and extensive evaluation of immunological responses by estimating antibody in serum and T-cell response of these proteins in future studies is required to understand their role in TB infection.
Footnotes
Acknowledgements
The authors are grateful to Director, National JALMA Institute for Leprosy & Other Mycobacterial Diseases (NJIL & OMD) for the support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Department of Biotechnology (BT/PR/6959/Med/14/913/2005), Government of India, New Delhi. CSIR-UGC provided a fellowship to GK.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.