
Abstract
Osteoarthritis (OA), the most common form of arthritis, causes pain and disability, as well as emotional distress. While total joint replacement is one of the most effective treatments available for improving the quality of life in people with severe OA, it is not suitable for all patients and all joints. Current pharmacological analgesics have limited efficacy, and their use is often restricted by adverse events. Medications that might reduce pain by slowing or preventing structural disease remain elusive. Our increasing understanding of the complex mechanisms that underlie OA pain offers a wide range of potential new treatment targets. New drugs for OA pain might come from repurposing those developed for other conditions, as well as novel compounds targeting pain mechanisms specific to the joint. Here we discuss the mechanisms of OA pain and its therapeutic implications. We explore evolving treatment modalities, including combination treatment. We review recent research and patents pointing to future OA therapies. We discuss the potential for biomarkers to facilitate drug development and targeting.
What Is Osteoarthritis?
Osteoarthritis (OA) is the most common disease of synovial joints, and a major source of pain, disability, and distress. Peak OA incidence is from the sixth decade, and with increasing longevity and employment into older age, OA has become an increasing burden on the economies and healthcare systems of the developed world. In the absence of a cure, OA increases in prevalence with increasing age, such that everyone who survives beyond the age of 70 might expect to develop OA in at least one joint. However, far from being simply an inevitable consequence of aging, OA might be better viewed as a heterogeneous group of related diagnoses, each displaying discrete genetic associations and precipitating and aggravating factors. For a majority of those unfortunate enough to experience end-stage disease, surgical arthroplasty might offer substantial pain relief and improve quality of life. However, arthroplasty is not currently possible for all joints, nor suitable or successful for all patients. The mainstays of treatment for most people with OA comprise analgesia and exercise. Existing drugs often offer little benefit, or risk important adverse events. Medications that might slow or prevent structural disease remain elusive.
OA has been defined both clinically and radiographically. Pain and crepitus are sufficient to make a clinical classification of knee OA in those aged over 40 years. 1 Radiographic characteristics evidence new bone formation (osteophytosis and subchondral sclerosis), cartilage loss (joint space narrowing), and subchondral cysts. Of these, osteophytes are most specific for OA, whereas joint space narrowing also occurs in other diseases, including rheumatoid arthritis (RA). However, the relationship between symptoms and radiographic appearance is only weak. Patients with advanced radiographic changes might be asymptomatic, whereas knee pain is common before radiographic features are apparent. Knee pain is predictive of subsequent radiographic OA development, 2 indicating a preradiographic stage in some patients, whereas for others, radiographic change precedes the onset of pain.
Attempts to define “early” OA have focused on the distinction from end-stage disease, in an attempt to identify subgroups of people for whom treatments that facilitate joint repair might offer secondary prevention. Tissue engineering approaches, for example, might eventually reduce disease burden and need for arthroplasty. However, the boundary between early OA and normality is not as easy to define. 3 Histopathological features, such as cartilage fibrillation and proteoglycan loss, might be seen in young adults, for example, those with chondromalacia patellae, but need not necessarily progress to OA in later life.
OA subgroups have been traditionally defined according to joint distribution (hip, knee, spine, and generalized nodal OA) or by predisposing factors (primary or secondary to injury or other joint disease). The impact of OA differs between different anatomical sites, with OA of large weight-bearing joints (hip, knee, and less often, ankle) often causing major disability, and OA of small joints of the hands impairing grip and fine manipulation.
Secondary OA offers potential for preventative treatments. RA in its early stages causes pain through immune synovitis, whereas secondary OA in later disease leads to persistent pain even after inflammation is fully suppressed. Immune modulation in early RA reduces joint damage as well as pain. 4 Optimal management of joint injuries focuses on the prompt relief of pain and functional restoration, and further research is required to optimize interventions that will prevent subsequent OA development. Reconstructive surgery for meniscal or cruciate ligament injuries has displayed limited capacity to retard the onset of posttraumatic OA, and current research focuses on the potential for tissue engineering approaches or medical interventions to improve long-term prognosis. 5
The contribution of OA to back pain remains controversial. Structural associations of low back pain remain even more difficult to define than in knee OA, with potential origins of pain in discs, as well as facet joints and periarticular structures.6,7 Radiofrequency denervation procedures designed to reduce nociceptive signals from facet joints have increasing evidence of benefit in carefully selected cases with low back pain, 7 although radiographic facetal OA is not predictive of pain outcomes. 6
Mechanisms of OA Pain: Therapeutic Implications
Biomechanical Model of OA Pain
Although OA has been recognized since ancient times, the mechanisms by which OA pathology causes pain have only recently been elucidated. Early perspectives viewed OA as primarily a mechanical condition of wear and tear, based on pathological cartilage loss and typical pain that is exacerbated by weight bearing and movement. Low muscle strength predicts worsening OA pain, 8 and muscle strengthening exercises can reduce OA pain. 9 This biomechanical model has led to treatments that offer relief to many, including exercise regimens, orthotics, and arthroplasty.
Pain improvement follows joint replacement surgery, although its precise mode of action remains uncertain. Arthroplasty removes diseased cartilage and subchondral bone, and reinstates a biomechanical and chemical barrier between synovial cavity and bone marrow spaces. Subchondral nerves are sectioned. By contrast, the synovium and periarticular structures remain intact or regenerate following arthroplasty. Synovitis might persist, but the foreign body reactions found postarthroplasty differ from the cellular and biochemical characteristics of the OA joint. 10 An unfortunate and important minority of people continue to experience significant pain after arthroplasty (estimated as 5% after total hip arthroplasty and up to 20% after knee replacement surgery), indicating either new pathological mechanisms consequent to surgery and prosthesis, or persistent OA pain mechanisms that are not resolved by arthroplasty.
Structural Associations of OA Pain
The biomechanical model led to extensive research being translated into clinical practice using imaging as a biomarker of OA. Radiographic joint space narrowing (
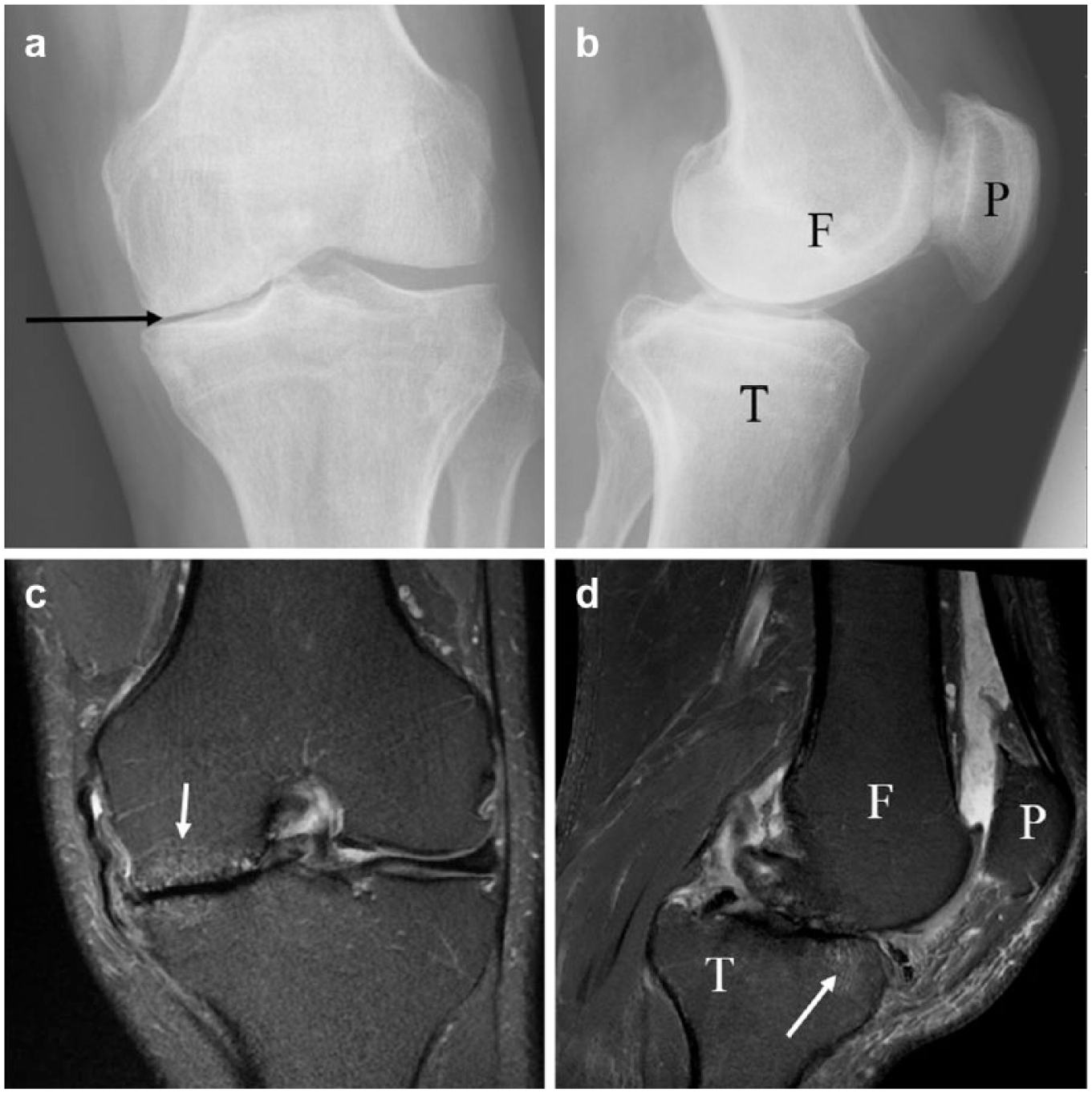
Radiographic and magnetic resonance (MR) images of the left osteoarthritic knee from a person with OA. The anterior-posterior (AP) projection radiograph (
Magnetic resonance imaging (MRI) has offered a more detailed assessment of the structural OA changes that might be associated with (and therefore mediate) pain (
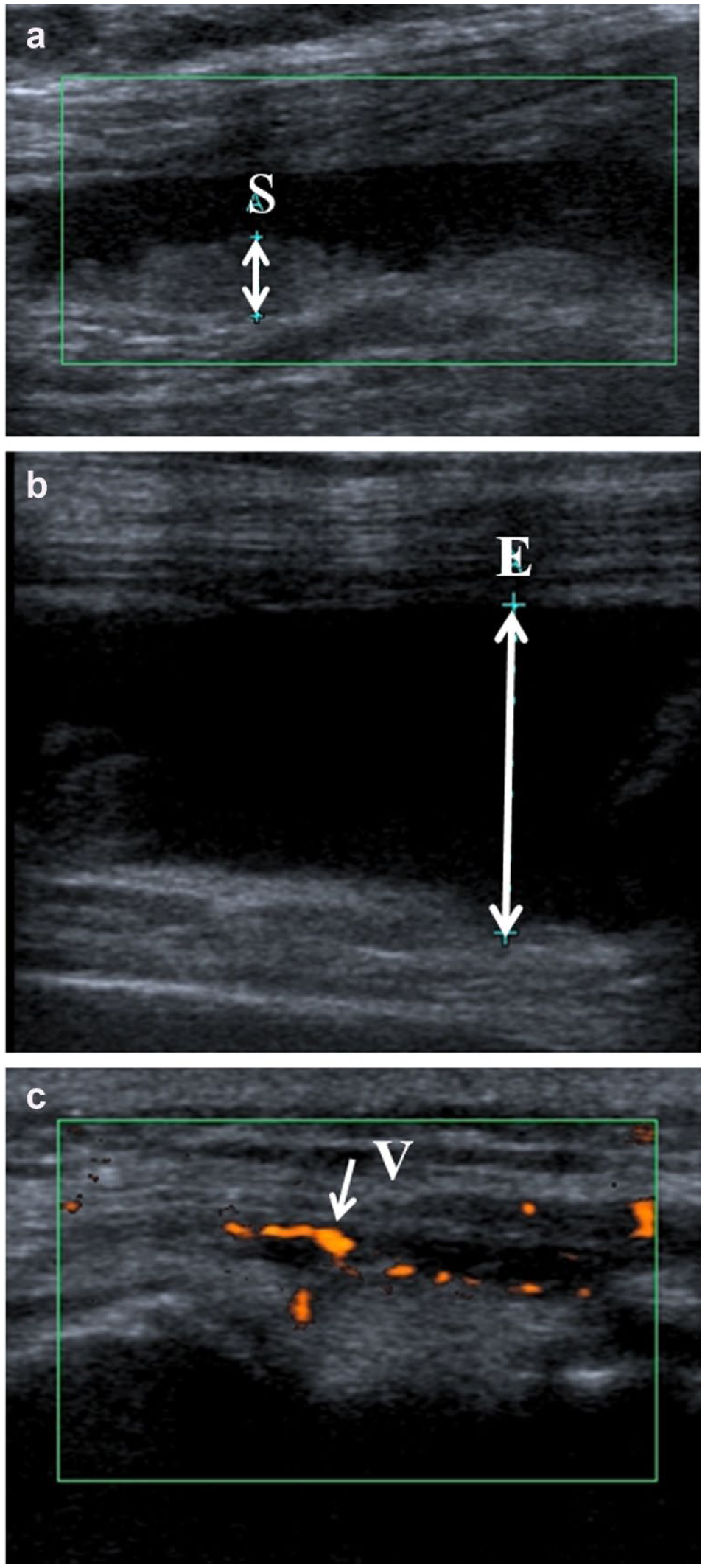
Ultrasound images of the knee of a person with OA: (
Current Limitations of Existing Therapies
Major limitations of existing therapies include limited efficacy, benefit for only a proportion of patients, the need for continuous treatment to maintain benefit and because of the waning effectiveness with time from treatment initiation, and potential for important adverse events. Nonsteroidal anti-inflammatory drugs (NSAIDs) can be helpful, but importantly, they increase risks of ischemic cardiovascular events and can impair renal function. Increased risk of gastrointestinal bleeding with NSAIDs requires coadministration of gastroprotective agents. Paracetamol has limited benefit and might itself be associated with gastrointestinal blood loss. 14 Opiates also have little sustained benefit for OA pain, and are associated with important gastrointestinal and cognitive adverse events, as well as their abuse potential. Local treatments, such as topically or intra-articular drug administrations, might circumvent some of these limitations, but even topical NSAIDs or capsaicin have limited benefits, and intra-articular treatments have limited duration of benefit, raising logistic issues for repeated administration.
There are no medical disease-modifying treatments consistently proven to reduce structural OA changes, and the most effective structural modifying treatment is surgical arthroplasty, which itself is expensive and might only have an 80% success rate in relieving knee pain. 15 Demonstration of disease-modifying activity in OA requires expensive and protracted clinical trials with large numbers of patients, due to the relatively slow and unpredictable rates of structural progression experienced by most people with OA, and the weak association between joint structure and clinically important outcomes, such as pain.
Limitations of preclinical models have also hampered the development of new medical treatments for OA pain. Animal models and behavioral tests used for developing OA analgesics are discussed in more detail elsewhere. 16 Rodent models of OA pain are useful for drug development, although large animal models might better reflect some the biomechanical influences in human OA. Pain behavioral techniques are well validated in rodent models, but more reproducible in rats than in mice, and more difficult in guinea pigs. OA is a disease of late maturity, and there remain concerns about the translational validity of induced OA in immature rodents. Surgical OA induction might better reflect initiating factors in at least some human OA (e.g., that following meniscal injury), whereas chemical induction, for example, by intra-articular injection of monoiodoacetate, provides a simpler method for rapidly developing a model of late-stage OA in rats. Different models might reflect different aspects of human OA, but there is no single model that reflects all aspects of human OA pain, and multiple models might need to be used to facilitate the generalizability of findings to human OA.
Modern Perspectives on OA Pain: The Quest for New Treatments
Current mechanistic understanding of the biochemical and cellular mechanisms of OA pain has led to optimism for the development of new and better treatments. More than 500 worldwide registered patents were filed between January 2014 and June 2016 related to OA therapies and biomarkers ( Table 1 ). The focus of the majority of these patents can be categorized into treating inflammation, bone and cartilage, or sensory nerves. Novel tissue engineering and cell therapies continue to aim to protect structural integrity. Additional proposed treatments include traditional Chinese medicine or dietary supplements, although these often lack clear biomedical mechanistic rationale.
Osteoarthritis and Biomarker Patents Registered between January 2014 and June 2016 Identified on https://worldwide.espacenet.com (June 2016) (further patent details can be found in the supplementary reference table).

ADAMTS5, a disintegrin and metalloproteinase with thrombospondin motifs 5; AMPA/KA, Ampa kainate subtype of the glutamate receptor; BMP, bone morphogenetic protein; CCDC, coiled-coil domain containing; CDK, cyclin-dependent kinase; CGRP, calcitonin gene-related peptide; CRAC, calcium-release activated calcium; EGF, epidermal growth factor; Fas, tumor necrosis factor receptor superfamily, member 6; FGF, fibroblast growth factor; Flt3, Fms-related Tyrosine Kinase 3; Fms, feline McDonough sarcoma; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICE, interleukin-1β converting enzyme; IFN, interferon; IK 1, inward rectifier current; IL, interleukin; IL-1-RA= interleukin 1 receptor antagonist; Kit, stem cell factor receptor; LNc RNA, long noncoding RNA; MMP, matrix metalloproteinases; MTF1, metalregulatory transcription factor-1; NGF, nerve growth factor; NSAID, nonsteroidal anti-inflammatory; PEDF, pigment epithelium-derived factor; PGE, prostaglandin E; RARB, retinoic acid receptor beta; RGD, Arg-Gly-Asp; TIMP, tissue inhibitor of metalloproteinases; TMCO3, transmembrane and coiled-coil domains 3; TNF, tumor necrosis factor; Trk, tropomyosin receptor kinase; TSPAN, tetraspanin; TPV1, transient receptor potential cation channel subfamily V member 1.
Inflammation and OA Pain
Synovitis in OA differs from that in RA, reflecting in OA predominantly innate rather than acquired immune responses, characterized by macrophage rather than lymphocyte infiltration. 17 Correspondingly, the cytokine profile from an inflamed OA joint differs from that seen in RA, although some characteristics are shared between diagnoses, albeit usually at a lower intensity in OA. OA and RA synovial fluids each contain increased concentrations of tumor necrosis factor (TNF) α and interleukin (IL)-1, as well as growth factors, including nerve growth factor (NGF), transforming growth factor (TGF) β, and vascular endothelial growth factor (VEGF). 18 Both display lipid-derived inflammatory mediators, such as E series prostaglandins. 19 Active RA is characterized by a systemic acute phase response, with raised erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Acute phase response is also increased in people with OA compared with nonarthritic controls, although usually only to levels within accepted laboratory normal ranges. 20 Synovial fluid and serum cytokine concentrations, and serum acute phase response in OA, have each been associated with OA pain intensity ( Table 2 ). 20 However, intercorrelation between inflammatory biomarkers prevents definitive elucidation of which specific markers might mediate OA pain.
Biomarkers of OA Pain, 2014–2016.
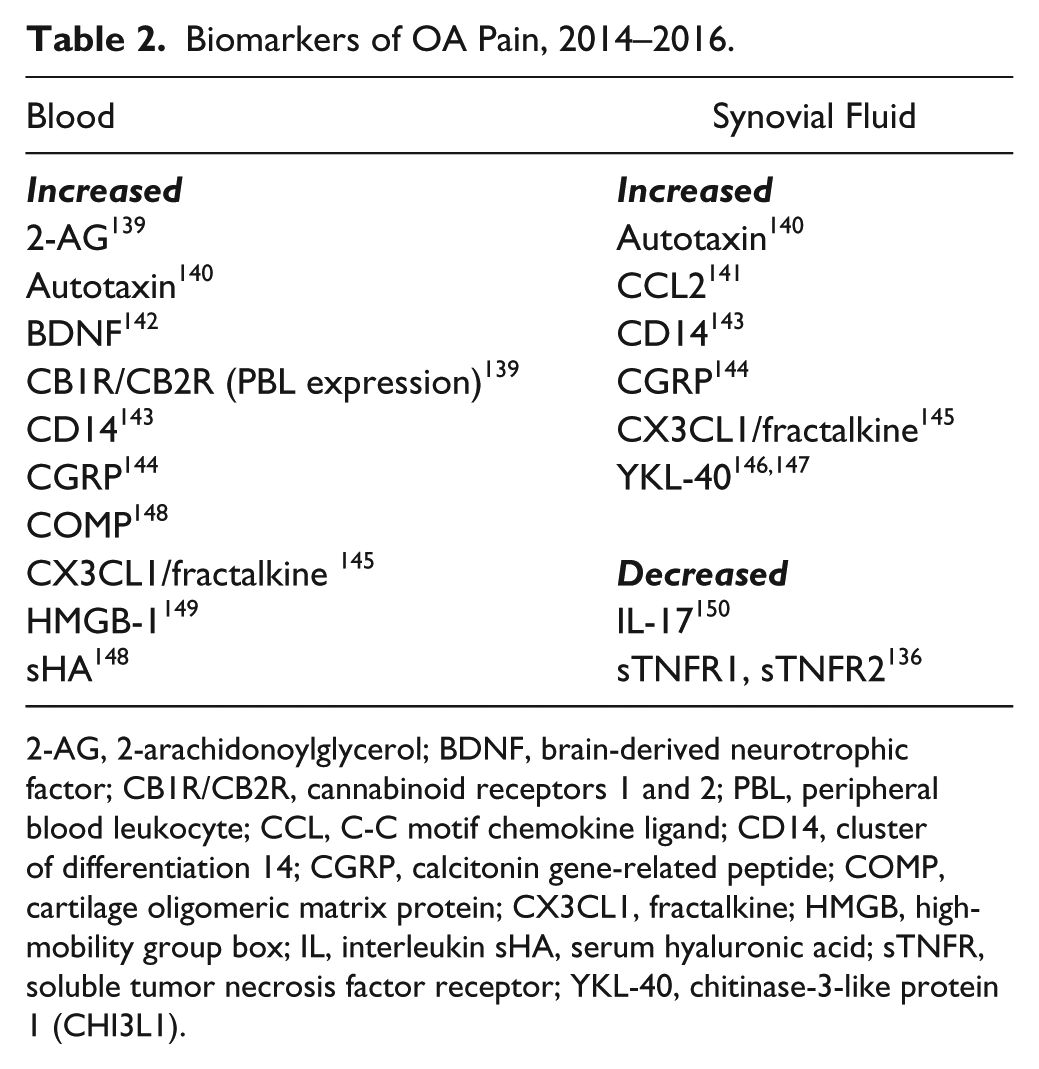
2-AG, 2-arachidonoylglycerol; BDNF, brain-derived neurotrophic factor; CB1R/CB2R, cannabinoid receptors 1 and 2; PBL, peripheral blood leukocyte; CCL, C-C motif chemokine ligand; CD14, cluster of differentiation 14; CGRP, calcitonin gene-related peptide; COMP, cartilage oligomeric matrix protein; CX3CL1, fractalkine; HMGB, high-mobility group box; IL, interleukin sHA, serum hyaluronic acid; sTNFR, soluble tumor necrosis factor receptor; YKL-40, chitinase-3-like protein 1 (CHI3L1).
Oral NSAIDs have long been known to reduce OA pain, and topical application might reduce pain from some arthritic joints, for example, hands or knees. 21 Glucocorticoids by local injection have analgesic benefit in knee OA, 21 but effects are temporary and repeated administration risks exacerbating structural damage, for example, through avascular necrosis. Systemic corticosteroids have not shown consistent benefit in OA, 22 and are largely precluded by adverse events with long-term use. Other drugs known to have anti-inflammatory disease-modifying activity in RA (hydroxychloroquine or methotrexate) offer some promise in OA, and the results of ongoing randomized controlled trials (RCTs) are eagerly awaited.23–26 TNFα is produced by synovium both in RA and in OA, and antibodies that block TNFα activity substantially reduce pain from active RA. 27 However, benefits have been inconsistent from TNFα blockade in OA,28–30 whether administered systemically or by intra-articular injection, and other inflammatory mediators might predominate in OA.
Recent research has further explored the potential of inflammation-related biomarkers to predict OA pain, including soluble CD14 and hyaluronic acid ( Table 2 ). Furthermore, inflammatory mediators are subject to intensive investigation as possible therapeutic targets that might both reduce pain and reduce inflammatory drive to joint damage ( Table 1 ). Recently patented agents have targeted IL-1, -17, or -18, and TNFα, as well as trying to improve targeting of more traditional OA treatment targets, such as prostaglandins and corticosteroid pathways.
Osteochondral Change and OA Pain
Subchondral bone is attracting increasing attention as a potential treatment target for OA pain. MRI-defined BMLs are associated with pain in cross-sectional and prospective studies. BMLs represent regions of increased metabolic activity and increased bone turnover, 31 and correspondingly, OA is associated with increased osteoclast activity within subchondral bone, both in man 32 and in animal models. 33 A range of osteoclast inhibitors have demonstrated analgesic efficacy in preclinical studies, including bisphosphonates, 34 osteoprotegerin, 33 and cathepsin K inhibitors. 35 Preliminary evidence from randomized clinical trials, which often focused on possible structural disease-modifying activity, suggest analgesic efficacy.36–38 Efficacy in unstratified cases with large joint OA was inconsistent, but based on the assumption that osteoclast inhibition should only be effective in the subgroup of people with OA who had increased bone turnover, Laslett et al. reported an RCT of zoledronate in people with knee OA and current BMLs on MRI scans at recruitment. 36 A single infusion of zoledronate was followed by significant pain reduction at the primary end point of 6 months. Another RCT, using neridronate, further suggests similar analgesic activity, 37 and a definitive trial of zoledronate is due to be completed in 2017. Strontium, which has mixed anabolic and anticatabolic activity on bone, also reduced BMLs 39 and showed pain-reducing effects in human OA, 40 although potential cardiovascular events might limit implementation of these findings into clinical practice.
Recent patents aiming to develop novel bone-targeted treatments might also realize the potential to reduce symptoms, while avoiding the need for prolonged and expensive clinical trials aiming to modify structural progression ( Table 1 ). Many of these agents, such as strontium, might also have structural effects mediated through chondrocyte function.
Mechanotransduction
OA pain is characteristically both intermittent and constant. 41 Intermittent and constant pain reflect discrete domains, as determined by Rasch analysis of questionnaire data, 42 and therefore are likely to be underpinned by discrete mechanisms. Intermittent OA pain is characteristically mechanically transduced, for example, on weight bearing or during walking, and mediated by fast-conducting myelinated nerve fibers. Precise molecular pathways of nociceptive mechanotransduction are incompletely understood, but likely involve ion channels, such as TRPA1, 43 short transient receptor potential channel 3 (TRPC3), 44 piezo 2, 45 and others. 46 Mechanotransduction also requires interaction between nerves and ancillary cells. 46 Inhibiting mechanotransduction as a means of reducing OA pain is an attractive proposition, commonly exploited by nonpharmacological interventions that redistribute mechanical stresses across the joint, for example, through orthoses 47 or by surgical osteotomy. 48 Pharmacological manipulation of mechanotransduction has presented more challenges to date. Mechanotransduction is also important for hearing and blood pressure control, and genetic deletion of TRPC3 44 or acid-sensing ion channel 2 (ASIC2) 49 mechanotransducing molecules results in hearing-impaired or hypertensive phenotypes, respectively. Identifying molecules with a specificity for nociceptive mechanotransduction, however, remains a desirable objective for pharmaceutical development.
Peripheral Nervous System
Nociceptive transmission, in either peripheral or central nerves, offers another potential target for treating OA pain. Intra-articular lignocaine injection can produce temporary analgesia, 50 presumably in part through nonselective sodium channel blockade (although also through placebo effects 51 ). Sodium channel subtypes involved in nociceptive pain include Nav1.7, and antagonists of Nav1.7 have displayed analgesic efficacy in preclinical models. 52 Clinical development of Nav1.7 inhibitors has, however, been limited by adverse events, including arrhythmia, seizures, and sedation, probably in part through cross-reactivity with other sodium channel subtypes (e.g., Nav1.6). Greater selectivity for Nav1.7, and peripherally restricted antagonists that less easily cross the blood–brain barrier are entering clinical development for neuropathic pain.
The clinical relevance of peripheral sensitization to OA pain has been confirmed by the analgesic efficacy of three different NGF-blocking antibodies (tanezumab, fulranumab, and fasinumab) in people with OA, 53 and additional NGF-blocking antibodies are poised to enter clinical development. 54 NGF pathways and neurotrophin receptors, such as tropomyosin receptor kinase (Trk) A, represent a high proportion of recent patents for novel OA treatments ( Table 1 ). Analgesia with NGF-blocking antibodies has been substantial (effects above placebo greater than observed with NSAIDs) and sustained during repeated administration over months. Efficacy has been demonstrated in knee 54 and hip OA 53 and also in low back pain, 55 a condition notoriously resistant to analgesic medications in RCTs and clinical practice. Other approaches to NGF pathway inhibition have focused on small molecular inhibitors of TrkA kinase. Development has been frustrated by lack of specificity for TrkA over other kinase receptors, but recent small-molecule allosteric TrkA inhibitors have been described with >100-fold selectivity for TrkA over TrkB and TrkC, and even greater specificity over other tyrosine kinase receptors. Preclinical data suggest that nonselective or selective TrkA inhibition can reproduce the analgesic effects of NGF-blocking antibodies 56 (compare efficacy with data in Xu et al. 57 ).
The precise molecular and cellular mechanisms by which peripheral sensitization contributes to OA pain remain unclear, raising the possibility that specific molecular targets other than TrkA might also generate effective analgesic agents. TrkA is coexpressed with transient receptor potential cation channel subfamily V member 1 (TRPV1) and calcitonin gene-related peptide (CGRP), on sensory nerves that also express CGRP receptors. NGF induces phosphorylation, increased expression, and thereby increased activity of TRPV1. 58 Small-molecule TRPV1 inhibitors did not display OA analgesic activity in RCTs, although adverse reactions, both on target (e.g., hyperthermia) and possibly off target (e.g., hepatotoxicity), might have precluded the use of doses adequate for clinically important pain relief. Further, TRPV1 inhibitors that are selective for sensory nerve function rather than thermoregulation continue in development. NGF also increases CGRP expression, which itself contributes to peripheral sensitization in rat OA models. 59 Small-molecule CGRP receptor antagonists were similarly limited by liver toxicity, but more recently, CGRP-blocking antibodies have demonstrated efficacy in RCTs as prophylactic agents against migraine, 60 although an RCT of one CGRP-blocking antibody in people with OA pain was disappointingly negative.
Central Nervous System
People with OA frequently display evidence of central sensitization: augmented central nociceptor activity in response to normal primary afferent inputs. Increased temporal summation (augmented pain intensity felt on repeated application of an equal-intensity stimulus), increased intensity and distribution of secondary hyperalgesia around a focus of primary insult, and augmented receptive fields and amplitude of flexor withdrawal reflexes each suggest spinal sensitization by analogy with similar phenomena observed in experimental animals.
61
Spinal sensitization is partly mediated by N-methyl-
OA pain phenotypes are in part dependent on the integrity of central inhibitory and facilitatory pathways originating above the spinal cord. People with OA frequently display impaired conditioned pain modulation (CPM), whereby a second painful stimulus fails to suppress pain intensity elicited by a test stimulus. Blunting of CPM might reflect impaired descending inhibition and/or increased descending facilitation. Descending control is, in part, mediated by serotonin 63 and dopaminergic 64 pathways, and serotonin/noradrenaline reuptake inhibitors are most likely to display analgesic benefit for neuropathic pain in those with impaired CPM. 65 Pregabalin, despite demonstrating benefit in other centrally mediated pain states, such as fibromyalgia, 66 displayed an absence of analgesic benefit in one RCT of people with hip or knee OA. 67 This might indicate that central pain augmentation is a major contributor to OA pain in only a minority of patients. Duloxetine, a selective serotonin/norepinephrine reuptake inhibitor (SSNRI), displayed significant analgesic activity in people with OA, 68 although overall effect sizes were small compared with placebo, again possibly reflecting response heterogeneity between individuals. Placebo analgesia might also offer the potential to activate endogenous analgesic pathways through psychological (e.g., cognitive) mechanisms. Indeed, placebo analgesic effect sizes exceed pharmacological effects in RCTs of most analgesic agents tested in OA. 69 If duloxetine and pregabalin share analgesic mechanisms with placebo, comparisons against placebos in RCTs might underestimate their full clinical potential.
OA pain is both a sensory and emotional experience, and negative affect, reflecting an overlapping construct of anxiety and depression, is strongly associated with musculoskeletal pain. 70 Indeed, brain functional and connectivity signatures of OA pain are localized to brain structures, such as the periaqueductal grey and anterior cingulate cortex, 71 regions known to be involved in mood disturbance.72,73 Negative affect is associated with impaired CPM74–76 and reduced (more sensitive) pressure pain detection thresholds, 77 in people with or without OA.
Modulation of anxiety or low mood, either pharmacologically or through cognitive behavioral therapy, has the potential to improve not only the emotional component of OA pain, but also the alterations in central pain modulation that can augment its sensory component.65,78 However, not all antidepressant or anxiolytic classes have demonstrated analgesic efficacy in OA. Early research suggested that depression in people without chronic pain might be associated with higher (less sensitive) pain thresholds, 79 but interactions between negative affect and chronic pain and a primary drive to pain sensitization from anxiety or depression remain to be fully elucidated. Strong associations between catastrophizing, 80 or fear avoidance beliefs, 81 and current or future joint pain 80 might suggest a primary association with anxiety rather than depression.
Ongoing nociceptive barrage is believed to facilitate and maintain central sensitization, although systemic or chemical factors might also contribute. 82 Several analgesic strategies have been found to reduce evidence of central sensitization, most notably joint replacement surgery. Widespread reductions in pressure pain detection thresholds 83 and impaired CPM,83,84 prominent in people with knee or hip OA, can each normalize after successful joint replacement surgery. However, associations between these clinical correlates of central sensitization and pain are, in part, genetically determined,63,85,86 and might therefore precede OA onset, indicating that nociceptive input is only one of several factors leading to central pain augmentation. Furthermore, an important minority of people continue to experience pain after arthroplasty (estimated at up to 20% after knee arthroplasty and 5% after hip arthroplasty) and continue to display evidence of central sensitization, 87 suggesting that once developed, central sensitization might become independent of ongoing nociception.
A direct contribution of synovitis to the development of central sensitization has been suggested by clinical and preclinical studies. MRI evidence of synovitis, but not BMLs, was associated with reduced pressure pain detection thresholds in one cross-sectional study of people with OA. 88 Widespread reductions in pressure pain detection thresholds have been associated with serum acute phase response in human OA.89,90 In rodent models of OA pain, synovitis is followed by evidence of central sensitization. 91 It remains possible that cytokines or growth factors, other than those classically recognized as driving inflammation, might generate central sensitization. The neurotrophin NGF increases brain-derived neurotrophic factor (BDNF) expression and central release by dorsal root ganglion (DRG) cells, contributing to central sensitization. 92 The potential of anticytokine treatments or neurotrophin blockade to inhibit the development or maintenance of central sensitization, particularly in patients with inflammatory OA, remains to be fully explored.
Glial cell activation in the spinal cord (first microglia, later accompanied by astrocytosis), and subsequently in the brain, develops in parallel to central sensitization in animal models of OA, and administration of glial cell inhibitors reduces sensitization. 93 Positron emission tomography scans using radiolabeled antagonist ligand to translocator protein (TSPO) suggest increased glial activity in brains of people with chronic low back pain, 94 as do increased cerebrospinal fluid (CSF) concentrations in OA or other musculoskeletal pain of molecules (e.g., ILs and cystatin C) known to be expressed by glial cells.95,96 Some existing analgesics might inhibit glial cell activity, 93 and novel, centrally acting analgesics that might reduce glial-mediated central sensitization have considerable potential for OA pain.
OA-specific drug patents lag behind the recent increases in understanding of central pain mechanisms in OA ( Table 1 ). These central pain mechanisms might not be OA specific, but rather shared, for example, with neuropathic pain. Analgesics under development for nonmusculoskeletal conditions might therefore help at least some people with OA in the future.
Systemic Influences on OA Pain
Systemic factors also contribute to OA pain. Structural and painful OA are associated with body mass index (BMI), and weight reduction is associated with reduced pain, although the effect size of dietary intervention is small. 97 Associations between BMI and OA pain might partly be explained by increased biomechanical stress, although associations between BMI and OA hand pain, 98 and indeed between BMI and non-OA pain (e.g., fibromyalgia 99 ), point to additional pain mechanisms. BMI and OA pain might be linked genetically, and adipocytokines circulating at higher levels in obese patients might contribute to neuronal sensitization.100,101 Identifying the mechanisms that link BMI to OA pain could lead to more refined and effective analgesic treatment than can realistically be achieved by dieting alone.
Treatment Strategies
Two areas of intensive research are the use of combination therapies and the development of robust biomarkers to help design and target effective OA pain treatment. Both have the potential to increase benefit from existing therapies and go hand in hand with the development of novel treatments.
Combination Therapies
Given the multiple contributing mechanisms in OA pain, it is unsurprising that no single treatment has proved efficacious for all patients. Combination therapy is the norm. Pharmacological interventions are typically delivered within the context of other treatment modalities, including psychological and physiotherapeutic approaches. Combination pharmacotherapy is commonly used, despite little robust evidence of greater benefit over monotherapies, and despite potential for drug interactions and adverse events.
Combination pharmacotherapy might most likely be effective where drugs act through discrete mechanisms. Combining two NSAIDs should have no greater than an additive benefit, and combining full and partial agonists might be expected to reduce benefit. Regular use of sustained-release or long-acting analgesics is recommended for chronic, constant pain. However, patients often choose to use analgesics intermittently, or use top-up analgesia to treat pain flares. People use analgesics prophylactically to facilitate activities that are anticipated to be painful. Caution is indicated, however, in ensuring that treatment combinations genuinely meet patients’ needs rather than concealing abuse of prescribed medications. Combination therapies might also incorporate nonpharmacological interventions. For example, effective analgesia might facilitate engagement in exercise, and analgesic drugs might therefore synergize with physiotherapy. All medications should be provided together with matched cognitive reassurance. 102
RCTs of combination therapy are difficult, expensive, and rare. An ideal trial design might require several parallel intervention arms in a double-dummy design: double placebo versus monotherapy A + placebo versus monotherapy B + placebo versus combination therapy A + B. Clinical practice tends to adopt a step-up approach, in which case the sequence of interventions requires randomization, further increasing trial complexity. RCTs testing combination pairs of acetaminophen (paracetamol), NSAIDs, and/or opioids have been reported, although clinically important benefits of combination over monotherapy with one or both components have not been consistently demonstrated. 103 Recent evidence that paracetamol might share cyclooxygenase inhibition with NSAIDs, as well as risk of gastrointestinal blood loss, 104 raises concerns about paracetamol + NSAID combinations.
Unexpected adverse events might result from pharmaceutical combinations. Phase III RCTs of NGF-blocking antibodies revealed rapidly progressive OA (RPOA) at increased frequency in people taking NSAIDs concurrently with NGF-blocking antibodies. 105 NSAIDs alone have been associated with RPOA, and retrospective analysis of trial data indicated a synergistic and dose-dependent effect of added NGF blockade. 106 The mechanism of this interaction remains uncertain, and both actions on bone turnover 57 and joint usage 107 in the context of analgesia have been proposed.
Monotherapies acting through discrete molecular mechanisms on central pain processing have demonstrated small clinical benefits in RCTs in OA, but it is unknown whether their combination with peripherally acting drugs would be more effective than either treatment alone. Combinations of duloxetine plus gabapentinoid in diabetic neuropathy have shown marginal possible benefits compared with either alone, 108 but similar combinations have not been reported in OA. Epidemiological evidence has suggested that concurrent treatment with beta-blockers, such as propranolol, is associated with lower reported pain and lower opiate use in people with OA. 109
Monotherapies might be tried before combinations, and treatments should only be continued if providing a benefit, and continued at the lowest effective dose in order to minimize risk of adverse events. Possible synergy between analgesic drugs, however, means that failure to detect a clinically important benefit from monotherapy might not exclude potential benefit in combination. A “real-life” trial protocol in which patients only progress to combination therapy if they display partial response to monotherapy might conceal synergistic benefit from combinations. Furthermore, failure to respond to one analgesic agent might predict failure to respond to a subsequent analgesic, most obviously where both act through the same mechanism. The converse observation, in that those who respond to one treatment might be more likely to respond to another in the same class, has informed flare RCT designs where participants are enriched with responders by identifying those whose pain flares following withdrawal of their regular analgesic. 110 However, psychological factors also contribute to therapeutic failure. When expectations of pain are incongruent with repeated experience, alterations in central pain processing can either augment or suppress pain. 111 Repeated exposure to ineffective treatments (especially where associated with adverse events) might create an expectancy of nonresponse, thereby blunting placebo and augmenting nocebo effects. A step-up approach might therefore lead to underestimation of benefit from combination therapies, and should ideally be tested in RCTs against concurrent or step-down approaches.
Biomarkers and Treatment Targeting for OA Pain
Biomarkers have potential utility to help diagnose patient subgroups, measure disease burden, define prognosis, and predict treatment outcomes (efficacy and adverse events). 112 Patients with highest disease burden, or whose disease is likely to get worse, stand to gain most from treatment, and are therefore likely to display the highest likelihood of benefit over risk. Several biomarkers have provided evidence of identifying those most likely to experience progressively painful OA, including collagen degradation products, urinary cross-linked C-telopeptide (uCTX) v II and uCTXIα, 113 and serum cartilage oligomeric matrix protein (COMP). 114 Overall however, the predictive value of existing biomarkers, either alone or in combination, is weak. Evidence that a biomarker might predict prognosis might currently be more useful in pointing to possible disease mechanism rather than indicating tools for stratifying treatments.
Given the complexity of pain pathways, their numerous modulating factors, and the clinical heterogeneity of OA, it seems likely that different treatments might be effective for different people, for different aspects of their pain problem or at different stages of their disease. Stratification of patients to select groups that are most likely to benefit from treatment has potential to improve success in clinical trials of analgesics with specific modes of action. Recruiting from homogenous groups of people who are most likely to respond to treatment permits increased statistical power, reduced trial size, and reduced exposure of those unlikely to respond to potential adverse effects of treatment. Offering to patients the treatments to which they are most likely to respond, at the earliest point in their care pathway, should reduce the suffering and disengagement inherent in cycles of ineffective treatment. However, individuals with OA might represent points on a continuum, each with a unique probability of response but without definable boundaries that would justify treatments being offered to (or withheld from) discrete patient groups.
Predicting treatment response from pretreatment characteristics is further hampered by the observation that only approximately 25% of overall treatment effect is attributable to specific pharmacological activities rather than placebo response. 115 Who will or will not benefit from a treatment might therefore be better predicted by determinants of placebo response rather than by matching the pharmacological target to an individual’s pain mechanism. Predictors of placebo response differ substantially from factors that predict pharmacological response, and include genetic, gender, and personality characteristics. Accumulating risk of pharmacological adverse events with long-term treatment of OA pain might not be justified if analgesic mechanisms are due to treatment context, particularly if placebo analgesia might be achieved by other methods without pharmacological risk. Pharmacological benefit to an individual should ideally be determined both in clinical trials and in clinical practice in order to justify progression to long-term treatment of chronic arthritis pain. Distinguishing pharmacological from placebo response, however, would require robust and validated biomarkers of treatment effects, or multiple crossover, double-blind, placebo-controlled trial designs. Such methodologies have generally not been translated into clinical practice, and rarely inform individual decision making on treatment continuation.
Clinical history and examination provides the cornerstone of patient stratification but has limited sensitivity or specificity for predicting treatment response for OA pain. Those with the most severe symptoms stand to gain most from treatment, and treatments with the highest risk or cost are generally reserved for those with the worst disease. However, where clinical risk factors for treatment outcome have been identified, their predictive value might not be sufficient to stratify treatment allocation. For example, high BMI predicts worse outcome from arthroplasty, but arthroplasty remains a cost-effective treatment option for those with high BMI. 14 Clinical examination displays only weak correlation with imaging evidence of synovitis, and poorly predicts response to anti-inflammatory treatments, such as intra-articular steroid injections. 116 Biomarkers that are more robust are required to inform clinical decision making.
Genetic Biomarkers
Genetic differences between individuals explain up to 40% of OA pain, 117 representing interactions between a large number of genetic loci, each alone contributing little predictive value. Targeted gene approaches have provided evidence for contributions from specific genes in general OA populations, and genome-wide association studies suggest additional, as yet uncharacterized, loci. Genetic risk factors for OA pain include risk of OA structural change, including bone and cartilage matrix integrity and shape, and inherited variability in pain processing. Inheritance segregates between hip, knee, or hand OA, highlighting constitutional risk factors for OA subgroups. Specific associations with allelic variation in Nav1.7 and TRPV1 ion channels, P2X7 purinergic receptors, catecholamine O-methyl transferase (COMT), and paired amino acid converting enzyme 4 (PACE4)117,118 might be shared with other chronic pain conditions, including neuropathic pain, and reflect heterogeneity in pain neurotransmission. Genetic variation might underpin differences in emotional 119 or descending pain modulation, 63 implicating, for example, serotonin transporter polymorphisms. Genetic variants also predict placebo responses, 120 analgesic (e.g., opioid) responses, or adverse events. 121 The use of genotypes (either individually or in combinations) to select people with favorable risk–benefit ratios for specific treatments remains in its infancy in OA. Examples in other branches of medicine have tended to be most successful with variants in single genes, as exemplified by the thiopurine methyltransferase genotype predicting toxicity risk from azathioprine. The polygenic nature of OA pain makes genotypic stratification more challenging.
Wet Biomarkers
Markers measured in biofluids have offered additional hope in subgrouping people with OA by pain mechanisms ( Table 2 ). Markers of synovitis have shown some promise, although systemic inflammatory responses are much less in OA than in RA. Any single, symptomatic joint might contribute little to circulating biomarker levels. Synovial fluid cytokine levels have been more often associated with OA pain than blood biomarkers, but synovial fluid volumes might be small, and aspiration might not be a practicable procedure for routine use in the clinical management of OA. ESR and serum CRP might each be increased in OA compared with nonarthritic controls, although usually still within the “normal” range. 20 Associations between serum’s acute phase response and OA pain might reflect a contribution from synovitis, but might otherwise be explained by known associations between CRP and high BMI. 122 Reduced leptin and IL-6 were associated with decreasing OA pain, in parallel to weight reduction following a behavioral weight management 123 intervention. Serum levels of various cytokines have also been associated with OA pain ( Table 2 ), mostly in cross-sectional studies. Increases in serum cytokine levels over time have less frequently been associated with increasing OA pain.
Serum or urine biomarkers of cartilage turnover, including COMP ( Table 2 ), have predicted structural and pain progression in OA.113,114 Serum biomarkers of osteoclast activity have been developed for partnering osteoporosis treatment. OA pain has been associated with increased urinary degradation products of collagen I, 113 the predominant bone collagen, although also a constituent of other connective tissues. Tartrate-resistant acid phosphatase type b5 (TRAP5b), released by osteoclasts during bone degradation, can be measured in serum by enzyme-linked immunosorbent assay. Serum TRAP5b was associated with subchondral osteoclast numbers in OA knees at arthroplasty, and, in a nonsurgical cohort, was associated with current OA pain and predictive of pain at follow-up. 124 Circulating bone and cartilage biomarkers, however, have displayed only weak correlations with current symptoms, perhaps reflecting origins from tissues in addition to those joints that are affected by symptomatic OA.
Imaging Biomarkers
Imaging biomarkers have thrown light on OA pain subgroups. Joint space narrowing on posteroanterior weight-bearing radiographs are associated with OA pain. 124 MRI scans also indicate associations between pain and cartilage thickness or surface area, and also with meniscal morphology.125-128 Each of these MRI features is associated with joint space narrowing on plain radiographs. Arthroplasty provides a mechanical and chemical barrier between joint and subchondral bone, and might be most effective when that barrier has been disrupted by late-stage OA. Indeed, people with less preserved radiographic tibiofemoral joint space are more likely to benefit from total joint replacement surgery. 130 Other mechanisms might drive pain in people with OA in whom joint space is preserved, mechanisms that might persist after arthroplasty. Up to 20% of patients continue to experience disabling knee pain after arthroplasty, and ongoing research is determining the extent to which this is due to pain mechanisms that persist despite surgical treatment (e.g., augmented central pain processing 87 ), or to sequelae of joint surgery (e.g., neuropathic pain from intraoperative nerve damage 131 ).
Imaging evidence of synovitis predicts response to intra-articular glucocorticoid treatment. The predictive value of imaging is weak, although better than evidence of synovitis from clinical examination. Reductions in synovitis, for example, determined by a reduced early enhancement rate (EER) on dynamic contrast-enhanced (DCE)-MRI, 132 can accompany pain reduction following steroid injection. MRI evidence of BMLs has been used to select patients for recruitment to trials of bisphosphonates, primarily on the basis that BMLs predict structural progression. Initial trials were designed to reduce structural progression by inhibiting subchondral osteoclast activity. Reductions in BMLs were accompanied by reductions in pain following intravenous zoledronate 36 or neridronate. 37 It remains unclear whether bisphosphonates would have demonstrated similar analgesic benefit in people without BMLs, although earlier RCTs of bisphosphonates in unselected patients with OA showed less consistent analgesic benefits. 133
Toward Patient Stratification
Biomarkers have potential to stratify patients, identifying those most likely to benefit from each of a range of treatments. However, further research is needed to determine which specific marker might best, and independently, predict treatment outcomes. Diverse biomarkers correlate with each other, and biomarker combinations might prove more specific and sensitive for detecting patient subgroups. For example, patients with a predominantly peripheral drive to their OA pain might display combinations of BMLs, synovitis, and loss of osteochondral integrity, while those with a major contribution for alterations in central pain processing might display changes in quantitative sensory tests, negative affect, and diurnal fatigue. Such biomarker combinations might cross traditional disciplinary boundaries, and it is currently little understood how wet biomarkers might correspond to imaging evidence of OA disease activity or, in combination, might increase their predictive value. 134 Some blood biomarkers of inflammation display only weak correlation with imaging evidence of synovitis by ultrasound or MRI, although it remains possible that more specific circulating cytokines might inform of the nature of imaging-determined synovitis, and thereby indicate specific treatment targets.
Patient stratification depends on a degree of phenotypic stability over a period of therapy. Defining phenotypes that remain stable over long periods approaches diagnostic classification. It is currently unclear whether synovitis reflects a stable OA phenotype or fluctuates throughout the disease. Latent class analysis of pathological samples has suggested that OA knees can be grouped largely according to the intensity of histological synovitis (scored based on hyperplasia and lymphoid infiltration), and that such groups are concealed among patients with similar clinical characteristics and demographics. 3 Generalized nodal OA has discrete genetic risk factors 135 and displays moderate synovitis, 136 further suggesting that inflammatory OA subgroups might be contained within the wider OA population. Synovitis has been identified in both early and established OA, and association with articular damage indicates a risk factor for structural progression, rather than a characteristic restricted to late-stage disease. 137 Synovitis might also be episodic, and BMLs are dynamic changes in OA, which might fluctuate from week to week. 138 Increases and decreases in synovitis or BML size correspond to changing OA pain, suggesting that pain might be an indicator of OA inflammatory or subchondral “disease activity.” Offering treatments during periods of active disease might be more acceptable to patients who might experience protracted periods of symptomatic remission, and might avoid risks from unnecessary treatment. Accessible and acceptable biomarkers that can define inflammatory, osteochondral, or neuronal mechanisms that underlie active disease could inform the management of OA flares.
OA biomarker studies remain in their infancy, having progressed from investigating biomarkers of structural disease to the clinically pertinent issue of OA pain. Key areas for future research will be to define those biomarkers alone or in combination that best predict the need for treatment or successful treatment outcome. RCTs of novel treatments aiming to benefit a subgroup of patients with OA will need to be paired with suitable biomarkers that will permit cost-effective treatment in clinical practice. Although biomarkers of structural disease might also predict pain outcomes, other biomarkers might await discovery that better predict pain than structural disease.
Summary
OA pain remains a major source of distress and disability, and is expected to increase as our populations continue to age. Current pharmacological analgesics have limited efficacy for what is typically a long-term condition, and their use is often restricted by clinically important adverse events. Our increasing understanding of the complex mechanisms that underlie OA pain offers a wide range of potential new treatment targets, and expectation is high that improved treatments will be available in the near future. New drugs for OA pain might come from repurposing those developed for other conditions (depression, anxiety, neuropathic pain, osteoporosis, and inflammatory arthritis), as well as novel compounds targeting pain mechanisms specific to the joint. Medication is unlikely to be a complete solution for people with OA, and even when pain relief is achieved, nonmedical interventions, such as physiotherapy, occupational therapy, orthotics, or psychological treatment, might be needed to restore normal function and well-being. Total joint replacement is one of the most effective treatments available in modern medicine for improving quality of life, and yet many people do not return to previously valued activities. New treatments are needed, and their development requires careful evaluation of their benefits and possible adverse events, within the context of the key needs of people with OA. A silver bullet is unlikely for such a heterogenous disease as OA, but new treatments will extend the benefit of existing therapies, while not necessarily replacing them all.
Footnotes
Acknowledgements
The authors gratefully acknowledge Mr Tom Kurien and Dr Aliya Sarmanova for the radiographic, MRI, and ultrasound images.
Abbreviations
2-AG 2-arachidonoylglycerol
ADAMTS5 A disintegrin and metalloproteinase with thrombospondin motifs 5
AMPA/KA Ampa kainate subtype of the glutamate receptor
ASIC2 Acid-sensing ion channel 2
BDNF Brain-derived neurotrophic factor
BMI Body mass index
BMLs Bone marrow lesions
BMP Bone morphogenetic protein
CB1R/CB2R Cannabinoid receptors 1 and 2
CCL C-C Motif Chemokine Ligand
CCDC Coiled-coil domain containing
CD14 Cluster of differentiation 14
CDK Cyclin-dependent kinase
CGRP Calcitonin gene-related peptide
COMP Cartilage oligomeric matrix protein
COMT Catecholamine O-methyl transferase
CPM Conditioned pain modulation
CRAC Calcium-release activated calcium
CRP C-reactive protein
CSF Cerebrospinal fluid
CX3CL1 Fractalkine
DCE Dynamic contrast-enhanced
DRG Dorsal root ganglion
EER Early enhancement rate
EGF Epidermal growth factor
ESR Erythrocyte sedimentation rate
Fas Tumour necrosis factor receptor superfamily, member 6
FGF Fibroblast growth factor
Flt3 Fms-related Tyrosine Kinase 3
Fms Feline McDonough Sarcoma
GM-CSF Granulocyte-macrophage colony-stimulating factor
HMGB High mobility group box
ICE Interleukin-1β converting enzyme
IFN Interferon
IK 1 Inward rectifier current
IL Interleukin
IL-1-RA Interleukin 1 receptor antagonist
Kit Stem cell factor receptor
LNc RNA Long noncoding RNA
MMP Matrix metalloproteinases
MTF1 Metalregulatory transcription factor-1
MRI Magnetic resonance imaging
NGF Nerve growth factor
NMDA N-Methyl-D-aspartic acid
NSAIDs Nonsteroidal anti-inflammatory drugs
OA Osteoarthritis
PACE4 Paired amino acid converting enzyme 4
PBL Peripheral Blood Leucocytes
PEDF Pigment epithelium-derived factor
PGE Prostaglandin E
RARB Retinoic Acid Receptor Beta
RCT Randomised controlled trial
RGD Arg-Gly-Asp
RPOA Rapidly progressive osteoarthritis
RA Rheumatoid arthritis
sHA Serum hyaluronic acid
SSNRI Selective serotonin/norepinephrine reuptake inhibitor
sTNFR Soluble tumour necrosis factor receptor
TGF Transforming growth factor
TIMP Tissue inhibitor of metalloproteinases
TMCO3 Transmembrane and Coiled-Coil Domains 3
TNF Tumor Necrosis Factor
TPV1 Transient receptor potential cation channel subfamily V member 1
TRAP5b Tartrate resistant acid phosphatase type b5
Trk Tropomyosin receptor kinase
TRPC3 Short transient receptor potential channel 3
TSPAN Tetraspanin
TSPO Translocator protein
TPV1 Transient receptor potential cation channel subfamily V member 1
uCTX Urinary crosslinked C-telopeptide
VEGF Vascular endothelial growth factor
YKL-40 Chitinase-3-like protein 1 (CHI3L1)
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplementary material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.