
Abstract
The aim of this study is to improve the aqueous solubility of a group of compounds without interfering with their bioassay as well as to create a relevant prediction model. A series of 55 potential small-molecule inhibitors of tumor necrosis factor–alpha (TNF-α; SPD304 and 54 analogues), many of which cannot be bioassayed because of their poor solubility, was used for this purpose. The solubility of many of the compounds was sufficiently improved to allow measurement of their respective dissociation constants (Kd). Parameters such as dissolution time, initial state of the solute (solid/liquid), co-solvent addition (DMSO and PEG3350), and sample filtration were evaluated. Except for filtration, the remaining parameters affected aqueous solubility, and a solubilization protocol was established according to these. The aqueous solubility of the 55 compounds in 5% DMSO was measured with this protocol, and a predictive quantitative structure property relationship model was developed and fully validated based on these data. This classification model separates the insoluble from the soluble compounds and predicts the solubility of potential small-molecule inhibitors of TNF-α in aqueous solution (containing 5% DMSO as co-solvent) with an accuracy of 81.2%. The domain of applicability of the model indicates the type of compounds for which estimation of aqueous solubility can be confidently predicted.
Introduction
A common issue in drug discovery is the low aqueous solubility of small-molecule drug candidates that in many cases precludes their evaluation by bioassay and results in their premature exclusion from further exploitation. This issue is most common in early-stage drug discovery, in which hits of low potency are obtained and higher concentrations are required to detect bioactivity. 1 With respect to potential inhibitors, the only exception to this rule concerns a minority of drug compounds with very low Kd and/or IC50 values (from low- to sub-micromolar levels). In this case, the concentrations needed for bioassay are below the limits of poor solubility (<20 µg/mL). 2 Low solubility is also an issue during the later stages of drug discovery, in which more than 40% of new chemical entities in the pharmaceutical industry have unacceptably low aqueous solubility. 1 Furthermore, according to Lipinski, 3 different discovery approaches, such as high-throughput screening (HTS) and structure-based drug design, adopted by two big pharmaceutical companies (Pfizer and Merck) have led in both cases to drug compounds with growing molecular weight and, in the case of HTS, poorer solubility. The latter can be explained by the good correlation of lipophilic substructures of the pharmacophores with enhanced activity 4 and has raised interest in studies devoted to the prediction of aqueous solubility of such compounds. 5 A knowledge of the aqueous solubility of drug compounds is crucial for data processing, too. In the case that solubility is incorrectly estimated, erroneous management of results and weak structure–activity correlations are highly probable. 5 Moreover, low aqueous solubility can lead to underestimated compound activity 4 as well as to nonspecific inhibition introduced by compounds, especially promiscuous ones, forming aggregates. 6
Tumor necrosis factor–alpha (TNF-α) has been associated with numerous autoimmune disorders such as psoriasis and Crohn’s disease, and it plays a pivotal role in the pathogenesis of rheumatoid arthritis. 7 SPD304 ( Fig. 1 , compound 1) is a small-molecule inhibitor of TNF-α that disrupts protein trimerization. 8
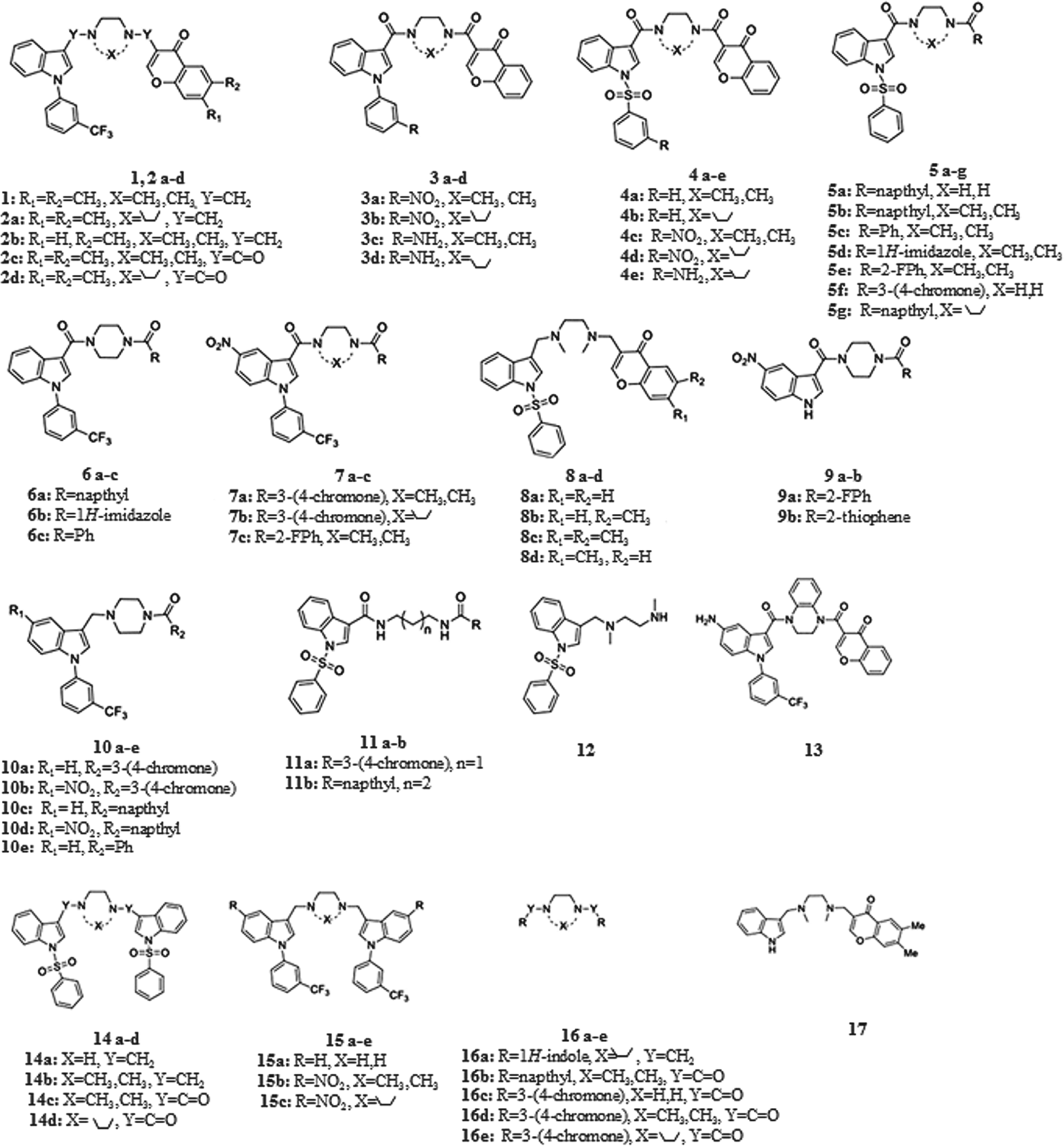
Structures of the 55 compounds studied (SPD304; compound 1 and 54 SPD304 analogues; compounds 2a-17).
When SPD304 enters the body, its 3-substituted indole moiety is dehydrogenated by the P450 isophorm, CYP3A4. Thus, a reactive electrophilic iminium intermediate is formed, which can potentially cause side effects by covalently binding to nucleophilic residues of protein and/or DNA. 9 Therefore, it cannot be used itself as an anti–TNF-α drug but only as a basis for drug discovery of new small-molecule inhibitors that disrupt TNF-α trimer formation. Thus, the development of disruptive TNF-α small-molecule inhibitors of relatively low toxicity remains a highly desirable goal.
Previously, we designed, synthesized, and biologically evaluated 38 SPD304 analogues.10,11 Herein, we have designed and synthesized 16 new compounds (
A major issue during the evaluation of these compounds was that many exhibited low aqueous solubility. This is always likely when the target site is hydrophobic, as is the case with TNF-α: TNF-α is a typical example of a protein with a shallow ligand-binding site that binds hydrophobic inhibitors. Indeed, SPD304, which has low aqueous solubility (10 µM in citrate/phosphate buffer, pH 6.5), 12 binds to the hydrophobic pocket of TNF-α by interacting with specific residues, the majority of which are hydrophobic ( Fig. 2 ). 8
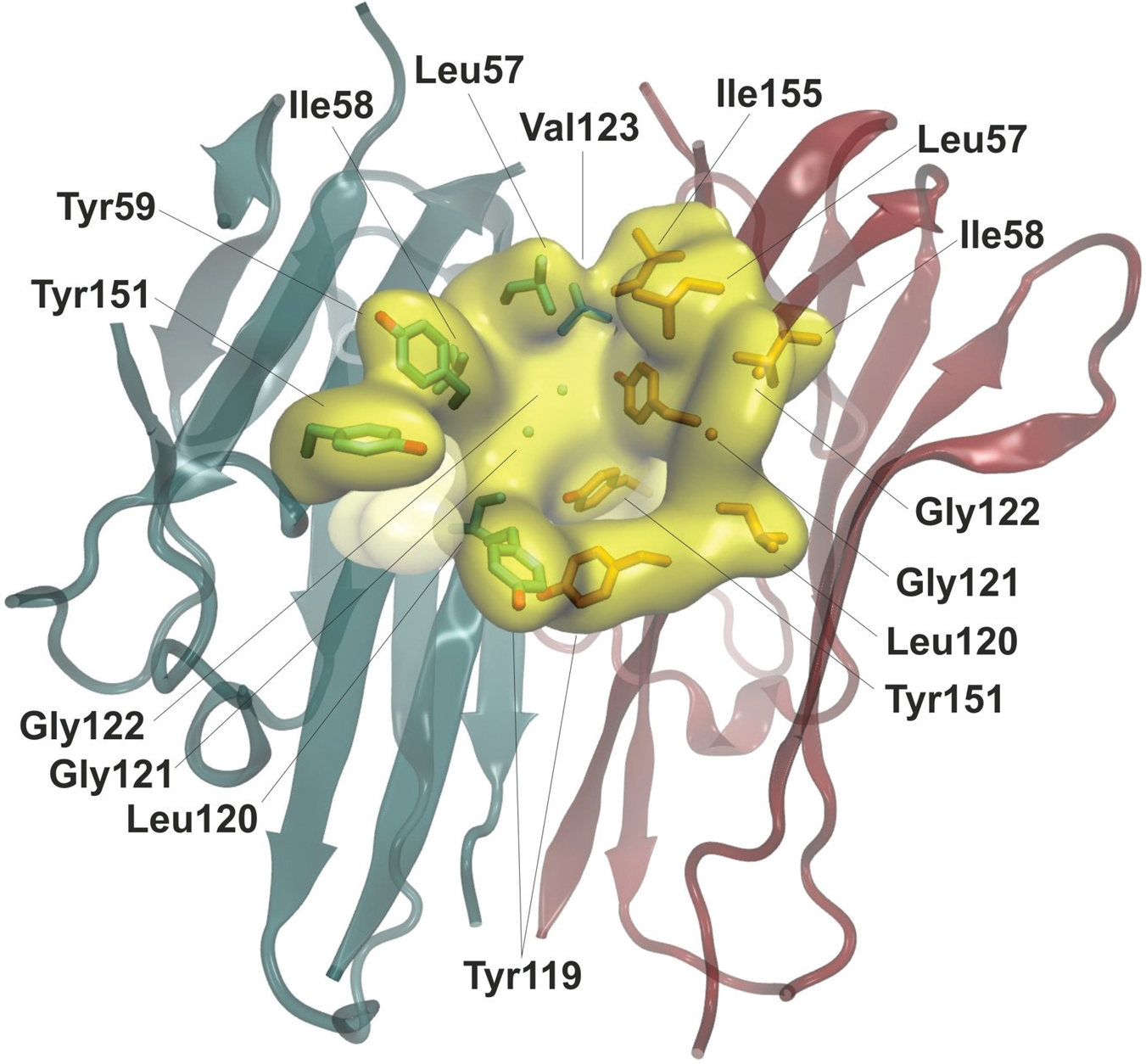
TNF-α has a shallow ligand-binding site where SPD304 binds to by interacting with specific residues, the majority of which are hydrophobic. Chain A: L57, Y59, S60, Q61, Y119, L120, G121, G121, Y155. Chain B: L57, Y59, S60, Y119, L120, G121, Y155.
Different dissolution protocols were evaluated, taking into account factors that influence aqueous solubility such as co-solvent addition, state of the solute before dissolution (solid/liquid), and dissolution time. Co-solvency is the most common approach in assay protocols regarding solubility enhancement. 13 Our study evaluated the impact on aqueous solubility on the addition of DMSO and PEG3350 at concentrations that do not hinder either protein activity or the biochemical assay.12,14
Quantitative structure property relationship (QSPR) models for the prediction of aqueous solubility can be based exclusively on the structural characteristics of compounds, in contrast to other types of such models, in which the knowledge of specific physicochemical properties, such as the dielectric constant or the solubility in pure solvent, and so forth, are needed. 15 The molecular descriptors encode information about the structure, branching, electronic effects, chains, and rings of the modules and thus implicitly account for cooperative effects between functional groups that limit or enhance aqueous solubility. Because poor pharmaceutical properties, and especially low aqueous solubility, contribute to increasing costs and time required for the development and formulation of drugs, such theoretical approaches are attracting growing attention.5,8,16 They can guide both medicinal chemists through the drug discovery process and biologists on the purchase of compounds 17 and the selection of co-solvents (a solvent added to an aqueous solution in ≤10%) for the optimization of bioassays. Several approaches exist for the prediction of aqueous solubility. Among these are the General Solubility Equation, 18 ESOL (Estimated SOLubility) method proposed by Delaney, 19 and the in silico prediction of aqueous solubility incorporating the effect of topographical polar surface. 20 These methods, among others, are not applicable in our study. Predictive models typically refer to either pure organic solvents 21 or pure aqueous solutions with no co-solvent added. 5 In the second case, predicted values are often lower compared with solubility values obtained after addition of co-solvent. Thus, prediction models based on data in which a co-solvent has been added 17 can increase the percentage of compounds predicted as soluble, proposing more compounds for further exploration. Moreover, another common disadvantage of currently available predictive models is the use of patchy solubility data retrieved from databases. Such data often present important discrepancies, not only in methods and protocols used but also in the definition of solubility, which can have a large impact on experimental values.5,8 Therefore, the creation of a QSPR model using high-quality and homogeneous experimental data generated with a consistent protocol and based solely on the compounds’ structural characteristics is desirable. 17 In our case, the low correlation between our experimental log S and calculated log D and clog P values ( Suppl. Fig. S1 ), which has been previously highlighted, 22 led us to the development of a validated QSPR model for solubility based on the findings of the present work. Our target was to discriminate the highly soluble compounds from the other two categories (low/medium solubility). For the extraction of safe conclusions, we have merged low/medium soluble compounds to have bigger sets for each solubility category. Based on the x-means clustering approach 23 included in Konstanz Information Miner (KNIME), 24 two clusters were formed for the discrimination between “low” and “satisfactory” solubility. The boundaries for each cluster are as follows: 5.3 to 52.3 µΜ for the “insoluble” class and 68 to 278.5 µM for the “soluble” class. Our experimental results and our in silico QSPR implementation could be useful tools for both aqueous solubility enhancement and prediction.
The factors that can influence solubility and that guided our investigations were the method used to determine the solubility, the dissolution time, the presence of a co-solvent, and whether the sample was filtered. To enhance the solubility, we used a buffered solution with a co-solvent. 1 The selected co-solvents (5% v/v DMSO or 5% w/v PEG3350) do not interfere with TNF-α and are compatible with our bioassay. 12 The dissolution procedure itself gives rise to two different types of solubility, 25 and these will affect the values obtained: when the compound is in the solid state and in saturation, a thermodynamic solubility is determined. The alternative, kinetic solubility, refers to saturated samples prepared from an initial wet stock in which the compound has been dissolved in pure organic solvent 25 and is often used in drug discovery where predissolution of compounds in DMSO is quite common.13,16 In both cases, the buffered solution used had a pH value of 6.5, which is the pH of solubility assays for oral drug candidates and reflects the intestinal environment. 26 Our approach was to enhance the aqueous solubility of the compounds, on the basis of the selected in vitro biochemical assays, and incidentally the chemical environment of the drug absorption site. Consequently, the number of compounds that could be studied increased, thereby providing an improved potential for the discovery of a novel hit.
Materials and Methods
Drug Synthesis
Synthesis of SPD304 Analogues
The general synthetic routes applied for 16 of the 55 compounds (compounds
Solubility Determination
Protocol of Solubilization and Solubility Measurement
Our new solubilization protocol was adapted to the in vitro biochemical assays that we wished to pursue and the relatively small quantities of the compounds available (~1 mg). The compound concentration was 0.3 mΜ, so the consumption was quite low (~0.5 mg/triplicate of measurements, Vsample = 1 mL). Temperature was at 25 °C in accordance with the protocol for the biochemical assay. According to our experimental data, at a concentration of 0.3 mM, the solute was in excess. Specifically, after separation of soluble from insoluble material, dissolution of the precipitate in 1 mL of methanol had a measureable absorbance (ca. ≥0.1). Dissolution time was selected at 10 h ( Fig. 3A ). At the end of the dissolution process, samples were centrifuged (15,000 × g, 30 min) to separate soluble and insoluble fractions. In some cases with a heavy precipitate, this step was repeated. After solubilization of the compound, solubility was measured with our previously published direct ultraviolet (UV) protocol. 12
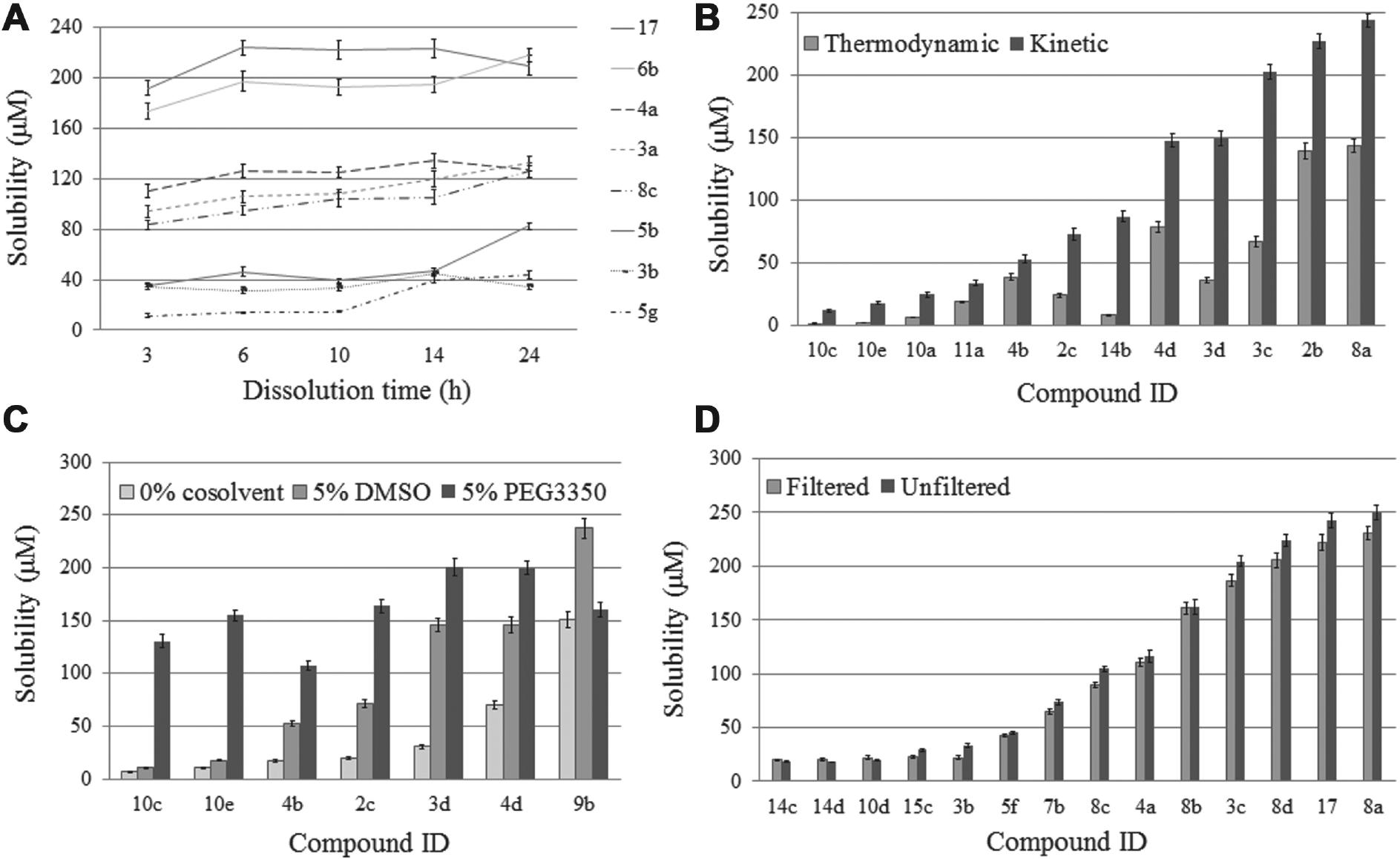
Study of some basic factors influencing the solubility of small-molecule inhibitors. (
Effect of Different Dissolution Time Intervals on Kinetic Solubility
Kinetic solubility ( Fig. 3A ) was determined as described below in 10 mM phosphate/citrate pH 6.5, 5% v/v DMSO, 0.3 mM of compound for different dissolution time intervals: 3, 6, 10, 14, and 24 h.
Comparison of Thermodynamic and Kinetic Solubility
Both thermodynamic and kinetic solubility ( Fig. 3B ) were determined in 10 mM phosphate/citrate pH 6.5, 5% v/v DMSO, 0.3 mM of compound, and dissolution lasted for 10 h.
Determination of Kinetic Solubility
Samples were prepared from an initial liquid stock of 10 mM of compound in 100% DMSO by dilution, which took place in sequential steps of addition: (1) stock of compound; (2) additional DMSO, until a concentration of 5% v/v was achieved; (3) 5× buffer solution (50 mM phosphate/citrate pH 6.5); (4) water. In this way, the compound precipitated less and kinetic aqueous solubility was enhanced.
Determination of Thermodynamic Solubility
The final buffer (10 mM phosphate/citrate pH 6.5, 5% v/v DMSO) was added to the solid compound to produce a final concentration of 0.3 mM. The solid compound was retrieved from a liquid stock in methanol by solvent evaporation. In this manner, the step of weighing was replaced by volume measurement, which allowed much smaller quantities to be used and experimental error to be minimized.
Effect of Co-Solvent Addition (5% DMSO, 5% PEG3350) on Kinetic Solubility
Kinetic solubility in 5% v/v DMSO, 5% w/v PEG3350, or without co-solvent (0% co-solvent; Fig. 3C ) was determined in 10 mM phosphate/citrate pH 6.5, 0.3 mM of compound after 10 h of dissolution.
Effect of Filtration on Kinetic Solubility
Kinetic solubility was determined in 10 mM phosphate/citrate pH 6.5, 5% v/v DMSO, 0.3 mM compound ( Fig. 3D ). After 10 h of dissolution, the soluble and the insoluble fractions were separated by (1) simple centrifugation (15,000 × g, 30 min) for “unfiltered” samples and (2) centrifugation (15,000 × g, 30 min) followed by filtration with inorganic membrane syringe filters (Anotop 10 IC, 0.2 mm, 10 mm) for “filtered” samples ( Fig. 3D ).
Proposed Solubilization Protocol for Bioassays
Τhe solubilization process (Scheme 1) starts with preparation of a stock solution of the compound (minimum 6 mM) in either 100% organic solvent (such as pure methanol) or 90% v/v DMSO/10% v/v water and proceeds with the dilution in a buffer with up to 5% co-solvent (0.3 mM of compound in 10 mM phosphate/citrate pH 6.5) followed by dissolution for 10 h. Soluble and insoluble fractions are separated by centrifugation, and solubility is measured according to our protocol. 12 The next step depends on the measured solubility; if it is sufficient for the selected biochemical assay, then the assay is performed; if not, an alternative co-solvent (e.g., 5% w/v PEG3350) appropriate for bioassays 12 is tried. If this too has failed, then a longer dissolution period is employed (≥15 h). For more details on this procedure, please refer to the supplemental material.
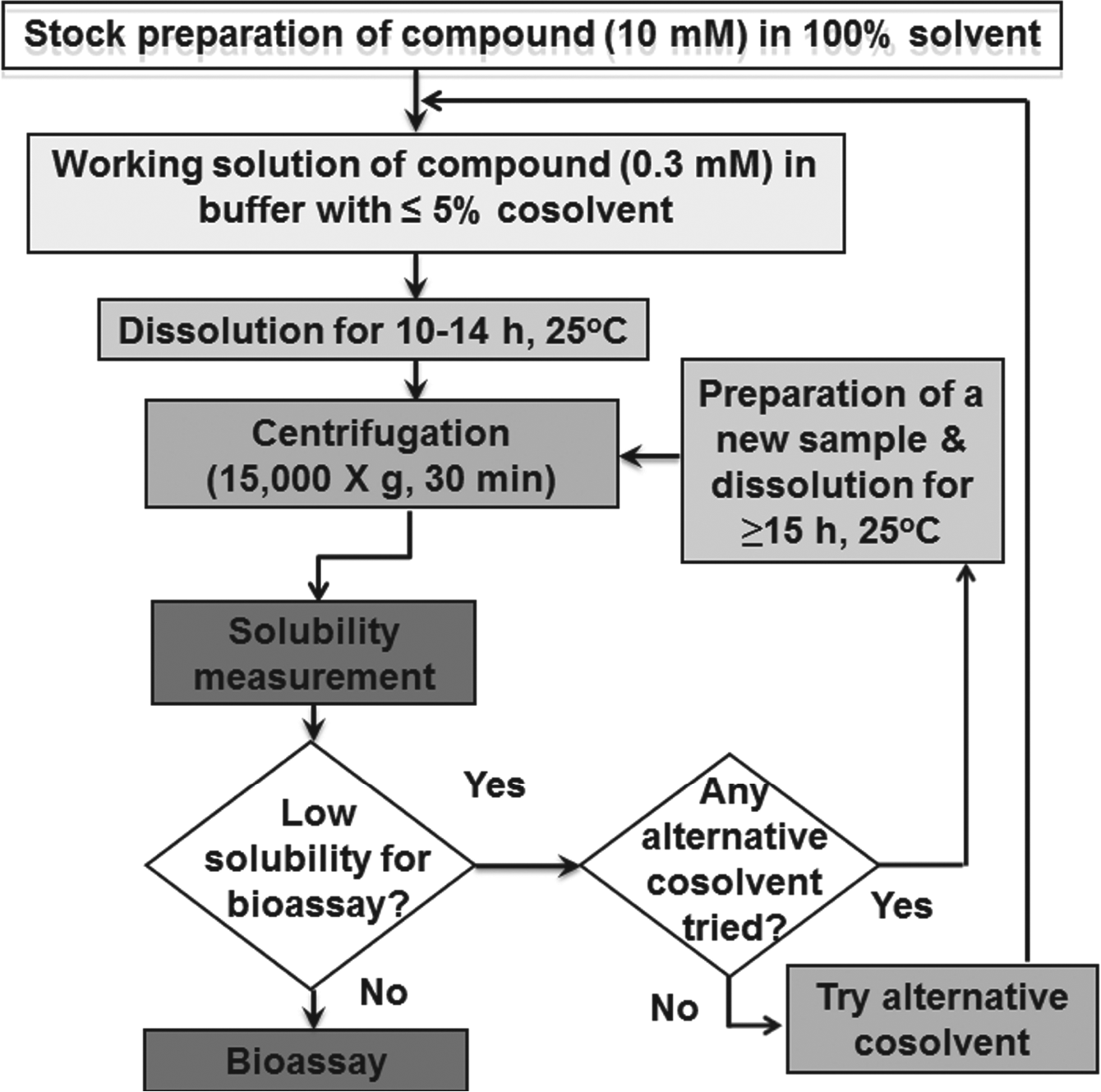
Schematic description of the procedure described at the proposed solubilization protocol.
All solvents used (DMSO, methanol, and PEG3350) were appropriate for UV spectroscopy. In all cases, samples were measured at 25 °C and immediately after the end of the solubilization procedure to avoid any additional precipitation.
Computational Analysis
Data Set: Descriptor Calculation
All structures for the 55 compounds herein were assembled in a single database, and their solubility values were classified into two categories: “soluble” and “insoluble” on the basis of the aqueous solubility measured in 5% v/v DMSO ( Suppl. Table S3 ). Mold2 software assessed the structural characteristics of the compounds used in this study based on a large and diverse set of molecular descriptors encoding two-dimensional chemical structure information. 27 Our in-house Enalos Mold2 KNIME node was used within our workflow for Mold2 descriptor calculation. Among the available descriptors, a filter was applied to remove those with no discrimination power. This resulted in a reduced set of 453 descriptors from the 777 initially available.
Model Development
Different variable selection and machine-learning methods can be applied in QSPR studies,28,29 and among these, the combination that best describes the correlations for a given data set needs to be explored. This task was facilitated by the KNIME platform that minimized the time needed to run and compare different methods in an effort to explore which of the available methods best described a given data set. The k-nearest neighbor (kNN) 30 method was selected over different methods tested within our workflow, as it outperformed (in terms of internal and external validations) all others tested. The method was chosen in combination with a variable selection technique. Variable selection techniques are needed in many chemoinformatics applications, and different methods have been successfully applied as variable selection tools in QSPR problems. Before running the modeling method, the most significant attributes among the 453 available were preselected for the training set using Best First variable selection and CfsSubset evaluator, which are included in WEKA. 31
Model Validation
To assess its predictivity, the model developed was fully validated both internally as well as externally, 32 paying special attention to the principles of model validation for accepting QSPR models as described by the Organisation for Economic Cooperation and Development.
The proposed classification models were validated using the following measurements: precision, sensitivity, specificity, and accuracy. The confusion matrix is also given.
External validation was applied by randomly splitting the data set into training and validation set in a ratio of 70:30. The separation of the data set was performed using the Kennard & Stones algorithm 33 included in Enalos+ KNIME nodes. 24 Compounds that constituted the test set were not involved by any means in the training procedure.
Domain of Applicability
The need to define an applicability domain expresses the fact that QSPRs are models that are inevitably associated with limitations in terms of the different types of chemical structures, physicochemical properties, and mechanisms of action for which the models can generate reliable predictions. The domain of applicability34–36 was defined using similarity measurements. Our in-house Enalos Domain–Similarity KNIME node was used to assess the domain of applicability of the proposed model. 24 First, similarity measurements defined the domain of applicability of the models based on the Euclidean distances among all training compounds and the test compounds. The distance of a test compound to its nearest neighbor in the training set was compared with the predefined applicability domain threshold. The prediction was considered unreliable when the distance was higher than this threshold. More information on the domain of applicability determination is given in the literature. 34
Results and Discussion
A direct UV method was chosen for solubility measurement because of its higher sensitivity compared with other methods, such as turbidity and nephelometry, which are not well suited for screening compounds of relatively low (<40 µM) aqueous solubility. 2 Previously, we developed a simple UV-based method (not requiring HPLC analysis) for the determination of aqueous solubility. 12
Four groups of compounds were created to study some basic factors that influence solubility and its measurement ( Fig. 3 ). The selection of the compounds in each group was made mainly according to cLogP and structure, so that each group contained compounds with low (≤4.2) and high (>4.2) cLogP and a variety of structural characteristics (linear/cyclic diamine bridge, different substituents, etc.). Furthermore, our results allowed us to establish a validated solubilization protocol for measuring the aqueous solubility of potential small-molecule inhibitors of TNF-α (Scheme 1) in the presence of 5% v/v DMSO. This protocol has been also applied to insoluble small-molecule inhibitors different from SPD304 analogues, for bioassays (unpublished data).
We studied the effect of dissolution time using eight compounds (
We then tested the effect of the initial state of a compound on aqueous solubility using 12 analogues (
Our previous studies indicated a variety of organic solvents suitable for the solubility enhancement of small drug molecules in bioassays.12,14 Based on these results, we examined the effect of selected organic co-solvents (5% DMSO, 5% PEG3350) on aqueous solubility. Both solvents are highly compatible with several specific protein assays,
14
including TNF-α binding assays.8,12 DMSO is widely used in the preparation of initial stocks of drug compounds.
25
Solubility of seven compounds (
The method of the separation of soluble and insoluble fractions may also have a significant impact on the measurement of solubility. This is possible for extremes in molecular properties; a strong association of compound molecules on the filter surface or floating on the sample surface (in the case of centrifugation of highly hydrophobic compounds) can give an adequate explanation for this phenomenon. 25 In our case, centrifugation was needed as our direct UV method also required the measurement of the insoluble fraction. 12 Comparison of equivalent data taken using centrifugation (unfiltered samples) and both centrifugation and filtration (filtered samples) revealed that the additional step of filtration after centrifugation did not significantly influence the measured solubility ( Fig. 3D ). As such, under the specific experimental conditions, it was not difficult to remove precipitant and potential micelles (bigger than 0.22 µm) by centrifugation, which was sufficient to effectively separate the two fractions.
Based on our results, we propose the solubilization protocol for bioassays in Scheme 1. The proposed actions for increasing the aqueous solubility of inhibitors for a successful bioassay are (1) predissolution of compounds in pure solvent; (2) addition of co-solvent such as 5% v/v DMSO, 5% w/v PEG3350, and so forth to the buffer 12 ; and (3) dissolution of compound under stirring for 10 to 14 h. In case that after this procedure a compound was not soluble enough for the bioassay, an alternative co-solvent can be tried such as glycerol, DMSO, or PEG3350. 12 For the same reason, the dissolution time can be increased to 15 to 19 h or more. The in vitro bioassay that follows solubility measurement can be spectroscopic, either UV or fluorescence, isothermal titration calorimetric, or, indeed, any other suitable method. It should be noted that the proposed percentage of co-solvent (up to 5%) in Scheme 1 concerns mainly in vitro assays, and it should be adapted to the appropriate bioassay accordingly. For example, in some cell-based assays, the tolerable percentage of co-solvent (i.e., PEG) may be 1% to 2%, 37 and it would be reasonable if a set of standard internal reference compounds were established as controls and provided acceptance criteria for the specific cell culture models. 38 The aim of the solubility measurement in this instance was to determine the exact concentration of the ligand at the start of the bioassay. In our studies, the bioassay that followed the solubility assay was based on fluorescence titration spectroscopy in chemical conditions identical to these of the solubility assay (10 mM phosphate/citrate pH 6.5, 5% co-solvent).
For building a predictive QSPR model, the 55 available compounds were classified into two broad categories, namely, “soluble” and “insoluble” ( Suppl. Table S3 ). We developed a predictive model using the KNIME platform (www.knime.org). To integrate and execute the different tasks within model development, we built a KNIME workflow suitable for data preprocessing, descriptor calculation, variable selection, modeling, validation, and domain of applicability determination. 24 More specifically, we integrated several existing KNIME nodes with our own in-house Enalos KNIME nodes that in combination can execute the following tasks: compound and solubility data preprocessing, Mold2 descriptors calculation and variable selection, kNN algorithm implementation, classification model validation, and the domain of applicability determination based on Euclidean distances.
The original data set of the 55 compounds was partitioned, based on the Kennard & Stones algorithm, into a training and validation set in a ratio of 70:30, consisting of 39 and 16 compounds, respectively. For each compound, 777 descriptors, which account for the topological, geometric, and structural characteristics, were calculated using the Modl2 Enalos KNIME node. 39 A filter was then applied for the removal of the descriptors that did not have discrimination power (values with no variation for more than 50% of the compounds). 40 In total, 453 descriptors remained to be used as possible inputs during the QSPR model development. The six descriptors selected as the most important for the development of the model are described in the supplemental material. A classification model has been developed to separate soluble from insoluble compounds. A kNN classification technique with five neighbors, implemented in the WEKA program, 31 was used to discriminate between the different classes. After the training of the classification model, prediction of the solubility of test compounds was performed.
The confusion matrix for the test set is presented in Table 1 . The performance of the model was evaluated according to the validation measurements already described. The significance, accuracy, and robustness of the model are illustrated by the corresponding statistics. By applying the model to the external test set, the following statistical results were obtained: precision = 80%, sensitivity = 88.9%, specificity = 71.4%, and accuracy = 81.2%. The applicability domain was defined for all compounds that constituted the test set (supplemental material). Because all validation compounds fell within the domain of applicability, all model predictions for the external test set were considered reliable (APD limit = 2.719).
Confusion Matrix of the Test Set.

To conclude, the following inferences can be made: (1) addition of 5% v/v DMSO or 5% w/v PEG3350 to aqueous solutions can significantly enhance solubility, (2) measurements using the thermodynamic protocol tend to produce lower solubilities than those from kinetic protocols, and (3) as filtration gives no reproducible differentiation in solubility over centrifugation alone, it is unnecessary for the separation of soluble and insoluble fractions. Moreover, we propose a new and validated approach for assessing solubilization of potential small-molecule inhibitors of TNF-α with reference to the measurement method used and the creation of a model for the prediction of solubility. The proposed protocol can help researchers enhance the solubility of their compounds and thereby prevent many from either being excluded from the evaluation process or being erroneously reported as “inactive.” It should be mentioned that in the current project, had the appropriate co-solvents not been used, some 90% of the potential inhibitors would have been identified as inactive as their aqueous solubility was below that required for determining inhibition. The objective was to find new hits with Kd < 20 µM, which can be a common indication of the discovery of a hit molecule. To determine a Kd of this value, solubility should be at least 40 to 50 µM under the experimental conditions of the bioassay. Solubility results in 5% DMSO or 5% PEG3350 could potentially apply to any compounds sharing physicochemical features with the above inhibitors and help researchers to eliminate the screening time of insoluble compounds, during biochemical assays. Finally, a validated QSPR model was created using the optimized solubility data in 5% DMSO ( Suppl. Table S3 ) and was effective in the prediction of aqueous solubility under these conditions. Because the design of new soluble molecules is based on the insertion, deletion, or modification of substituents at different sites of the molecule, this model could assist researchers in this procedure. The simplicity of the proposed approach makes it broadly applicable to virtual screening and data mining to identify soluble molecules.
Because of its high predictive ability and simplicity24,39,40 this work can be a useful tool for the selection of candidates for costly and time-consuming organic synthesis as well as for aqueous solubility enhancement of potential TNF-α small-molecule inhibitors. Thus, this prediction can be a guide toward the design and synthesis of promising compounds.
Footnotes
Acknowledgements
We are grateful to Dr. Campbell McInnes, South Carolina College of Pharmacy, and Professor Lindsay Sawyer, Edinburgh University, for English-language editing of the article.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the project TheRAlead (09SYN-21-784) co-financed by the European Union (European Regional Development Fund) and Greek national funds through the Operational Program “Competitiveness & Entrepreneurship,” NSRF 2007–2013 in the context of GSRT-National action “Cooperation.” The authors declare no competing financial interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.