
Abstract
Three-dimensional (3D) in vitro tissue models provide an approach for the systematic, repetitive, and quantitative study of drugs. In this study, we constructed an in vitro 3D acrylated hyaluronic acid (AHA) hydrogel model encapsulating fibroblasts, performed long-period 3D culture, and tested cellular topological changes and proliferation variation in the presence of herpes simplex virus-1 (HSV-1) as an infecting virus and acyclovir (ACV) as the treatment drug. The AHA hydrogels were formed by using Michael addition chemistry of bis-cysteine containing MMP-degradable cross-linker onto AHA prefunctionalized with cell adhesion peptides (RGD). Cellular structures of 3T3 fibroblasts in hydrogel presented different morphological evolution processes and proliferation rates between different groups, including HSV-1 treated alone, ACV treated alone, HSV-1 and ACV cotreated, and control samples. In AHA hydrogel, ACV blocked HSV-1 infection/replication on fibroblasts. Yet, the proliferation of ACV-treated fibroblasts was slower than that of the control group. A significantly longer period was required for cells in 3D AHA gel to regain a healthy status when compared with cells in two-dimensional (2D) culture. This hydrogel-based 3D culture model potentially lays a foundation for analyzing the response of self-organized 3D tissues to viruses and drugs in a way that is closer to nature.
Introduction
Tissue culture models in vitro based on cellular self-organization in the extracellular matrix (ECM) are preferred as experimental models to screen drugs and study disease,1–6 because the use of animal models in scientific studies may present issues with higher cost and ethical concerns. Commonly used two-dimensional (2D) cell culture models contributed to biological studies and other disciplines. However, we may overlook key phenotypic and functional characteristics when using this “flat biology” approach, and the results may not be predictive of in vivo tissue responses.3,7,8 2D monolayers often lack the physiological relevance of the tissues that are encountered by a pathogen during natural infection in vivo. Thus, only utilizing 2D cultures to predict the infection process is not sufficient. Therefore, to overcome some of the inherent limitations associated with the 2D monolayer cultures indicated above, extensive research efforts have moved to three-dimensional (3D) cell culture models.
The 3D cell culture model provides biomolecular cues that mimic the environment of natural tissues and promote cell-to-cell communication and cell-to-ECM interactions.8–10 Self-organized multicellular structures that emerged in 3D have the advantage of recapitulating the morphological structure of native tissues. Here the self-organized multicellular structures are defined as cell cultures formed by the growth of cells without external action or control in a specific way. Furthermore, the self-organizing process of cells in 3D multicellular models also mimics the fundamental biological processes related to virus invasion and drug treatment observed in vivo. Therefore, the 3D cell culture is a promising model to duplicate the in vivo environment of native tissues, crucial for achieving effective tissue repair, and to study the disease progression and the response of self-organized tissues to drugs.11–13 Currently, hydrogels, water-swollen, cross-linked polymer networks, have emerged as particularly promising materials for 3D cell culture models. The engineered hydrogel matrices3,14,15 provide a physiological context that closely mimics the natural ECM and can act as 3D scaffolding materials for testing biologically active and cell modulating substances. Therefore, events such as intercellular communication, cell–matrix interactions, and multicellular response to virus infection and drug treatment can be recapitulated in defined 3D hydrogels.16,17 Fibroblasts are the most common cells in animal connective tissues, and can be cultured for many research fields,18,19 including hydrogel-based 3D cultures. Fibroblasts can synthesize the ECM and collagen, the structural framework (stroma) for native tissues, and play a critical role in wound healing. Thus, it is important to study infected fibroblasts and drug treatment in an elaborately constructed culture model. Herpes simplex virus (HSV) is a human pathogen that infects its host via mucosal surfaces or abraded skin, 20 and the underlying dermis consists primarily of fibroblasts. Therefore, the study of HSV-1 infection on fibroblasts is important for advancing the research on drug therapy of HSV-related diseases. HSV-1 has been intensively studied due to its prevalence in human diseases, 21 and acyclovir (ACV) is a commonly used virus-specific drug for HSV-1. Therefore, HSV-1 and ACV were chosen as the tested virus and drug to demonstrate the performance of the 3D hydrogel-based culture model. Through the 3D culture model, the effect of the virus and drug at certain concentrations was evaluated in a 3D ECM that closely mimicked the in vivo environment.
In this study, we constructed an in vitro 3D acrylated hyaluronic acid (AHA) hydrogel model encapsulating fibroblasts, evaluated the cellular 3D pattern formation, and tested 3D cellular topological changes in the presence of HSV-118,21 as an infecting virus and ACV as the treatment drug. Herein, AHA was modified to contain acrylate functionalities using mild chemistry, which can be further used as cross-linking sites to form hydrogels. HSVs have been intensively studied due to their prevalence in human diseases.18,21 Fibroblasts were chosen as the study subject because they are among the most common cells of the connective tissue in animals and can synthesize the ECM and collagen. This elaborately constructed 3D hydrogel-based culture model serves as a suitable platform for studying 3D structure formation by fibroblasts under viral infection and drug treatment.
Materials and Methods
Cell Culture
NIH 3T3 fibroblasts (ATCC CRL-1658, Manassas, VA) were cultured on a 150 × 25 mm Falcon Plastic Petri Dish (Fisher Scientific, Waltham, MA, cat. no. 353025). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Fisher Scientific, cat. no. MT-10-013-CM) + 5% fetal bovine solution (FBS, Fisher Scientific, cat. no. MT35015CV) + 1% Penstrep (Fisher Scientific, cat. no. SV3008201) in a 37 °C chamber filled with 5% CO2. Cells were plated at a density of 2 × 105 cells/plate and were trypsinized every 3 days. In this study, the passage number of 3T3 fibroblasts was P20–P24. To harvest cultured cells from the substrate, the cell culture medium was aspirated and replaced by 5 mL of 0.25% trypsin-EDTA (1×) solution (Fisher Scientific, cat. no. 25200056). Cells were incubated with trypsin-EDTA (1×) solution for 5 min in the incubator mentioned above and detached from the cell culture plate. Detached cells were transferred into a 50 mL Falcon tube (Fisher Scientific, cat. no. 339652), centrifuged, resuspended in 1× fresh cell culture medium, and counted with a hemocytometer calculator (Fisher Scientific, cat. no. Z359629).
Preparation of HSV-1
HSV-1 KOS strain was a gift from Dr. William Halford (Southern Illinois University School of Medicine, Springfield, IL). 22 HSV-1 was harvested from Vero cell lines. A monolayer of Vero cells was first seeded on a 150 × 25 mm Falcon Plastic Petri Dish. One million copies of HSV-1 were then added to the cell culture medium. The infected cells were transferred into a 37 °C chamber filled with 5% CO2. Cells, as well as supernatants, were collected 3 days after infection. Both cells and supernatants were transferred into 50 mL Falcon tubes. After centrifugation, the supernatant was pooled into separate Falcon tubes. Cell pellets were resuspended with 1× phosphate-buffered saline (PBS) solution. In order to collect viruses left inside the cells, an alternative shock-frozen process was applied to the cell pellets by repeating the following process three times: freezing the cells at −80 °C for 10 min and then thawing them at 37 °C in a water bath for 10 min. The viruses released from the cells were then pooled together with the viruses harvested in the supernatant. In order to concentrate the virus, the pooled solution was centrifuged at 3500g at 4 °C for 30 min. Plaque assay was then used to count the final viral load.
Dilution and Application of Drug and Virus
ACV stock solution was prepared in DMSO at a concentration of 4 mg/mL and stored at −20 °C. The ACV stock solution was then diluted using regular cell culture medium to a concentration of about 10 µg/mL. The HSV-1 stock solution was kept at −80 °C at a concentration of 6.25 × 106 pfu/mL. It was then diluted by using regular cell culture medium to reach the final concentration. A multiplicity of infection (MOI, number of infectious viruses per cell) of 0.2 was used throughout the study, unless otherwise indicated. In order to infect 3T3 cells, HSV-1 was added to the cell culture medium for 2D monolayer culture or 3D hydrogel culture. The infected cells or gel samples were transferred into a 37 °C chamber filled with 5% CO2. For the 3D group with virus infection, the culture medium with virus was added into the wells containing cell-encapsulated gels on days 0 and 1 with MOI = 0.2. The medium was replaced by fresh medium every 2 days from day 2, without virus after the AlamarBlue assay. On other days, the culture medium was harvested and stored in sterile wells before the AlamarBlue assay, and then pipetted back into the wells again after the AlamarBlue assay was performed. For the group cotreated with virus and drug, the medium containing HSV-1 and ACV drug was added to the wells containing the cell-encapsulated hydrogel that had already gelled, and the plate was incubated at 37 °C with 5% CO2. The medium was replaced by fresh medium every 2 days from day 2. On other days, the culture medium was harvested and stored in sterile wells just before the AlamarBlue assay, and then pipetted back to the samples in the wells after the AlamarBlue assay, as described above. The virus was added to the medium only on days 0 and 1, and the drug was added at days 0, 2, and 4. For the group with drug alone, the medium containing ACV but without any virus was added to the cell-encapsulated gel samples, and then the plate was put into the incubator. The medium was replaced with fresh medium very 2 days. The drug was also added at days 0, 2, and 4. On other days, the culture medium was changed only for AlamarBlue assay. The medium was carefully replaced to avoid any breaks in the gels, and the sample should always be kept under sterile conditions.
Hyaluronic Acid Modification
Sodium hyaluronan was modified to contain acrylate functionalities. In this study, the hyaluronic acid (HA) modification involved a two-step process ( Fig. 1 A,B ). Briefly, HA (2.00 g, 60 kDa, 5.28 mmol carboxylic acids) was reacted with 36.8 g (211.1 mmol) adipic acid dihydrazide (ADH) at pH 4.75 in the presence of 4.00 g (20.84 mmol) 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) overnight and purified through dialysis (8000 MWCO) in deionized (DI) water for 1 week with changing water frequently. The purified intermediate (HA-ADH) was lyophilized and stored at −20 °C until used. All modified HA-ADH was reacted with N-acryloxysuccinimide (NHS-AC) (4.46 g, 26.4 mmol) in HEPES buffer (10 mM HEPES, 150 mM NaCl, 10 mM EDTA [pH 7.2]) overnight and purified through dialysis in a 100–0 mM salt gradient for 1 day, followed by dialysis in DI water for 3–4 days before lyophilization. The degree of acrylation was about 15% using 1H nuclear magnetic resonance (NMR) (D2O) by taking the ratio of the multiplet peak at δ = 6.2 (cis- and trans-acrylate hydrogens) to the singlet peak at δ = 1.6 (singlet peak of acetyl methyl protons in HA).
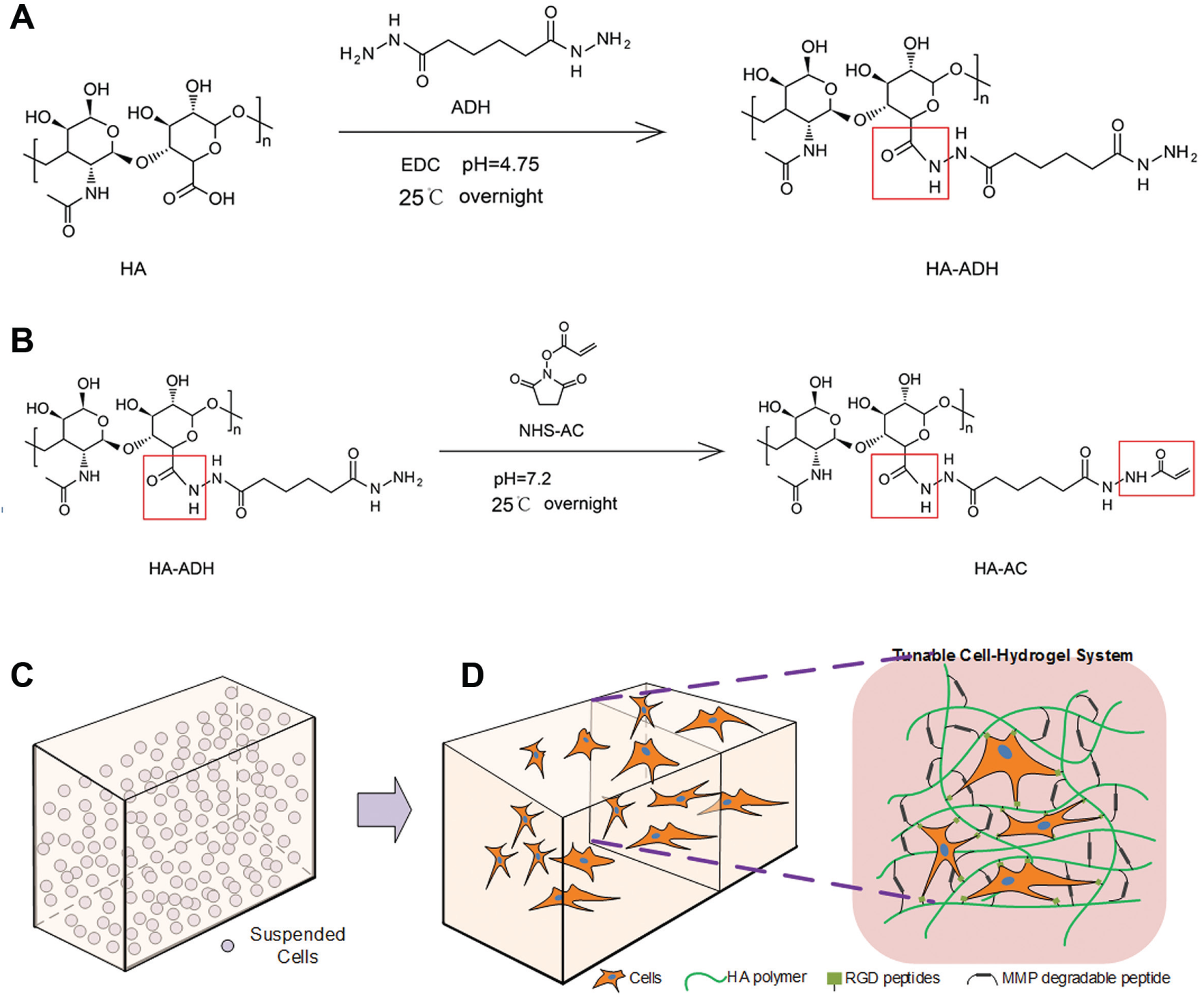
Schematic representation of the synthesis of HA-AC (
Cell-Laden HA Hydrogel Formation
HA hydrogels were formed by Michael addition of bis-cysteine-containing MMP-degradable cross-linker onto HA-AC prefunctionalized with the Ac-GCGYG-RGDSPG-NH2 adhesion peptide (RGD, Genscript, Piscataway, NJ). The schematic representation of the cell-laden HA hydrogel formation is presented in Figure 1C , D . Eight milligrams of lyophilized HA-AC was dissolved in 100 μL of 0.3 M TEOA buffer, mixed with the RGD peptides (0.1 mg/vial) in TEOA buffer, and placed for 25 min at 37 °C for the production of HA-RGD. Fibroblast single-cell suspension was prepared and placed on ice. One milligram of lyophilized cross-linker was then dissolved in 20 μL of 0.3 M TEOA buffer (pH = 8.4) or 0.1 M HEPES buffer and mixed with HA-RGD (final concentration 100–150 μM RGD) and cell suspension. The gel precursor solution was pipetted drop-wise (10 μL/drop) onto a sigmacote glass slide, then clamped with another sigmacote slide with plastic coverslip spacers, and finally incubated for 30 min at 37 °C to allow gelation. The final gel was swelled in culture medium before being placed inside the wells of 96-well plates for long-term culture. The mechanical properties of the hydrogels were controlled by varying the HA concentration, and the parameter r, which is defined as the mole ratio of thiol (–SH) in the cross-linkers to the acrylate group (–ACs) in the HA-ACs.
Rheology Measurement for Characterizing the Stiffness of Hydrogels
The storage (G′) and loss modulus (G′′) were measured with a plate-to-plate rheometer (Physica MCR, Anton Paar, Ashland, VA) using an 8.0 mm plate under a constant strain of 1% and a frequency ranging from 0.1 to 10 rad/s. The hydrogels were cut to a size of 8.0 mm in diameter. A humid hood at 25 °C was used to prevent the hydrogel from drying.
Cell Proliferation Measurement
The AlamarBlue (Life Technologies, Grand Island, NY) assay was used to quantify the relative metabolic activity of the cells inside the hydrogels, which could be used to measure the cell proliferation over time. 23 In this assay, 15 μL of AlamarBlue dye was mixed with 100 μL of phenol red free DMEM and added to each gel-containing well in the 96-well plates and incubated at 37 °C with 5% CO2 for 4 h. Following incubation with the reagent, the fluorescence signal was quantified using a plate reader with an excitation wavelength of 550 nm and an emission wavelength of 590 nm. The proliferation rates are presented as fold increase over the value obtained on the first day. Acellular hydrogels were used to adjust for background fluorescence. The experiments were performed by fabricating three gels for each sample type. Moreover, the whole experiment (in triplicate) was repeated four times in order to confirm the reproducibility of the measurements.
Results and Discussion
Preparation and Characterization of the Hydrogel for 3D Culture
Acrylates were conjugated onto the HA backbone through a two-step process ( Fig. 1A , B ). The modified HA hydrogel scaffolds were synthesized to contain different amounts of HA and cross-linkers (condition A, 3.0% HA, 100 μM RGD, r = 0.75; condition B, 3.5% HA, 100 μM RGD, r = 0.5). For these HA hydrogels, the HA backbone was modified with RGD peptides to introduce integrin binding sites on the cells encapsulated in hydrogels ( Fig. 1C , D ). RGD adhesion peptides were incorporated through Michael addition of the cysteine side chain in the peptide to the acrylate groups on the HA backbone. The RGD concentration influenced both the starting time and the extent of cell spreading. The cells started to spread earlier when using HEPES buffer than when using TEOA buffer in the course of making gel. Moreover, the extent of cell spreading increased with the RGD concentrations. However, overdosed RGD induced lower stiffness of the hydrogel, resulting in a faster degradation of the hydrogel, probably because the excessive RGD molecules conjugating to the –ACs from the HA-ACs decreased the statistical chance for cross-linkers to conjugate to the –ACs, which may not be beneficial to long-term 3D culture of cells in gel. The duration during which HA hydrogels could maintain their original shape also depended on mechanical properties. The mechanical properties of the hydrogels can be controlled by changing the HA concentration or the cross-linking density, r, which is defined as moles of –SH from the cross-linkers over moles of –ACs from the HA-ACs. Usually, higher HA concentrations or the cross-linking density will increase the time duration before the hydrogel collapses, which is beneficial to long-term culture. Therefore, a compromised lower concentration (100 μM) of RGD and adequate amount of the cross-linker (r = 0.5–0.75) were applied in the following experiment.
Impact of the Component Ratio on the Stiffness of Hydrogel
The storage (G′) and loss modulus (G′′) of the formed hydrogels were measured with a plate-to-plate rheometer. Results showed that the G′ and G′′ did not cross at any measured frequencies (0.1–10 Hz), and that the frequency independence ( Fig. 2A , B ) was consistent with typical hydrogel characteristics. The hydrogels were highly elastic, because the loss tangent values (averaged ratio of G′′ to G′) were lower than 0.1 for the hydrogels tested in this study. The mechanical properties of the hydrogels can be regulated by altering the HA concentration or the cross-linking density, r, defined as moles of –SH from the cross-linkers over moles of –ACs from the HA-ACs (refer to Fig. 1D ). The combinational effect of HA concentration, cross-linking density, and RGD concentration moderately determines the mechanical property of hydrogel. Different combinations of parameter values lead to different mechanical properties of hydrogel, as shown in Figure 2 . Nevertheless, there was no large difference in the mechanical properties between these two cases in Figure 2 (case A with 3% HA, 100 μM RGD at r = 0.75; case B with 3.5% HA, 100 μM RGD at r = 0.5). The 3D culture model should be elaborately constructed so that it can mimic the in vivo multicellular structure conditions. We chose the parameter values in these two cases empirically according to our previous research, 15 in order to achieve a balance between the cell growth rate and gel degradation rate.
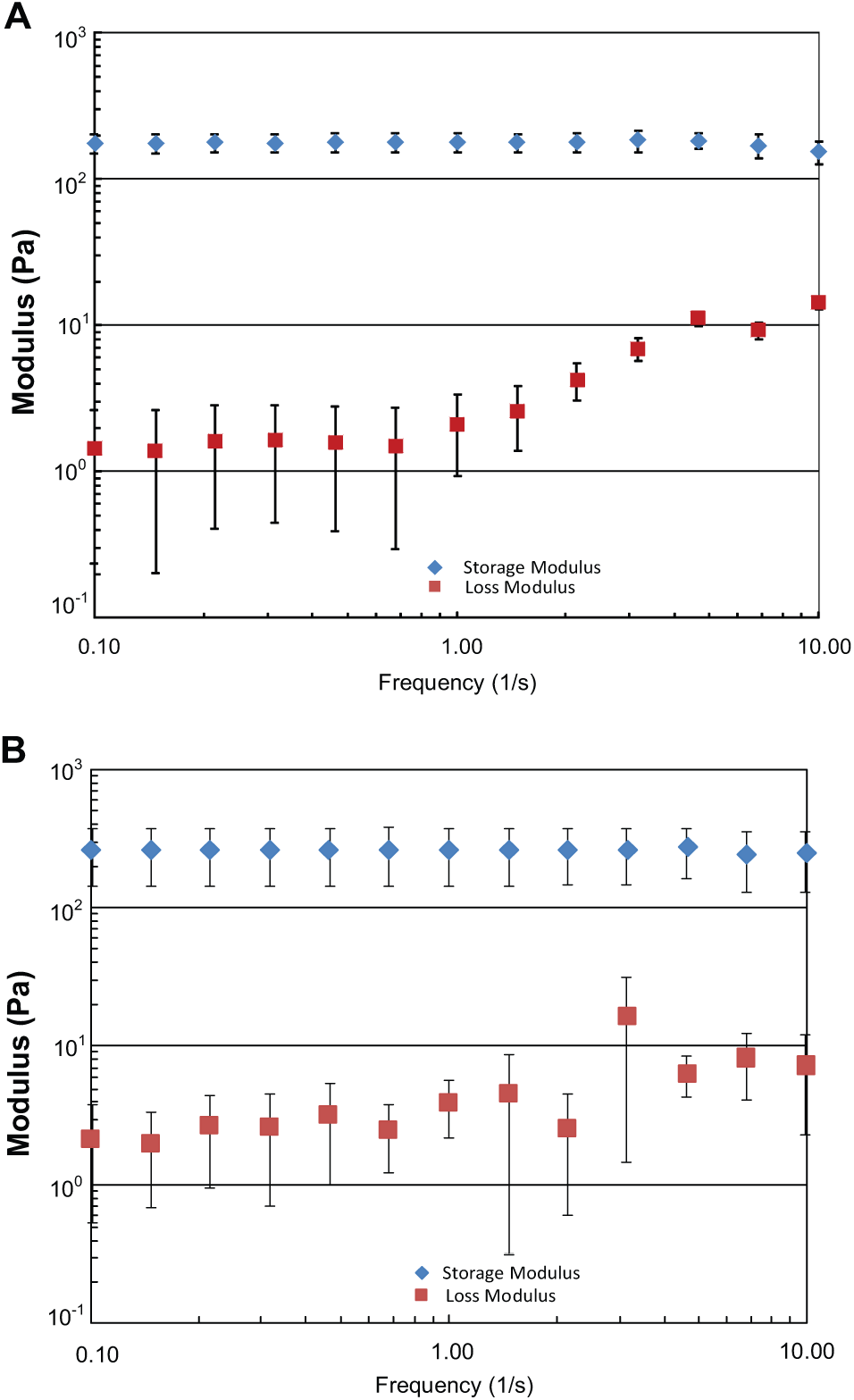
The storage (G′) and loss modulus (G′′) of HA-AC hydrogels. (
Morphology of 3D Multicellular Structure Formed by 3T3 Fibroblasts
3D cell culture models often promote levels of multicellular organization not possible in conventional 2D culture systems, 7 which provides an approach that bridges the gap between traditional cell culture and animal models.24,25 By constructing an in vitro 3D hydrogel model encapsulating fibroblasts and by performing the long-period culture, we tested the effect of HSV-1 as an infecting virus and ACV as the treatment drug. The cellular structures of 3T3 fibroblasts in the 3D AHA hydrogel presented a morphological evolution process after infection with HSV-1 and treatment with ACV. As shown in Figure 3 , the cells in the control group sprouted, spread, and connected to each other during the first 3 days, and got significantly denser at day 5. The group with infected cells in the AHA hydrogel exhibited slower growth and much less spreading than the control group. Infected cells treated with ACV showed slower cellular growth and a subhealthy condition with less intercellular connections than the control cells. However, these cells went through the recovery process after day 3 compared with the infected cells. To investigate the impact of the accumulation of ACV on cellular growth, the fibroblasts in AHA gel were treated with ACV alone as a separate group of experiments, as shown in Figure 3m–p . Their morphological evolution process was similar to that of the control group, but cells presented slower sprouting and spreading and started to connect to each other from day 3. The final density of cell networks ( Fig. 3p ) seemed sparser than that in the control group. In 2D cultures, the virus-infected culture presented a significantly different morphology compared with that of the control group at both 12 and 24 h, as shown in Figure 4a–d . ACV treatment alone did not induce obvious differences in cellular morphology when compared with normal cells ( Fig. 4e ). The time at which cells in the 3D AHA gel started to recover to a healthy status was significantly longer than that in 2D culture, since the cells in 2D can recover to normally spread and connect with each other after only 24 h, as shown in Figure 4f .

The cells inside the AHA hydrogel treated with the virus alone, drug alone, or with both the virus and drug. (

Morphology of 3T3 fibroblasts in 2D culture. (
Cell Proliferation in AHA Hydrogel
Proliferation within gels was measured by AlamarBlue assay. By using the REDOX indicator resazurin (oxidized form), the cellular proliferation can be spectrophotometrically measured in AlamarBlue assay. Viable cells continuously convert resazurin to resorufin, increasing the overall fluorescence. Thus, the alteration in the number of metabolic active cells can be approximately detected by measuring changes in the fluorescence of the dye in the intracellular environment.
To further characterize the proliferation of NIH 3T3 cells under HSV-1 and ACV treatment inside the modified HA hydrogels, AlamarBlue assay was performed for four groups of hydrogel-based experiments. For each group, two types of hydrogels (A and B), consisting of different composition proportions, as indicated in Figure 2 , were tested. The growth of 3T3 fibroblasts cultured in AHA hydrogel without any treatments in the control group is presented in Figure 5A . The cell proliferation showed different tendencies for hydrogel A and hydrogel B, as shown in Figure 5A . 3T3 cell proliferation was stronger in hydrogel A than in hydrogel B, mainly because hydrogel A had a lower stiffness than hydrogel B. The cells were allowed to proliferate for more than 6 days. Interestingly, the number of living cells in HSV-1-infected hydrogel samples increased from day 1 to day 3, but steeply decreased after day 3, as shown in Figure 5B . The decrease of living cells indicated that cells died over time after day 3, according to the fluorescent detection of cellular metabolic activity by AlamarBlue assay. The overall metabolic activity in the HSV-1-infected cells in hydrogel ( Fig. 5B ) was lower than that in the control sample shown in Figure 5A , especially after day 3. The cell proliferation results indicating the effect of ACV on the infected cells are shown in Figure 5C . The cellular metabolic activity continued to increase for at least 4 days before reaching a plateau between days 4 and 6, whereas the living cell amount moderately declined after day 6. The decline curve in Figure 5C after day 6 is much slighter that that in Figure 5B after day 3, probably indicating the cure effect of ACV upon HSV-1 infection in 3T3 cells in the 3D model. However, the slightly declined curve in Figure 5C may also imply the possible accumulative side effect of ACV, if we compare the overall cell metabolic activity with that in Figure 5A . Thus, an experiment evaluating ACV effects on normal 3T3 cells was also performed and cellular proliferation was measured ( Fig. 5D ). A tendency similar to that of the control group was observed, but a slightly lower proliferation rate was detected. This indicates that the accumulative side effect of ACV is small and the slightly declined curve shown in Figure 5C may be associated with the fact that ACV starts to lose its antiviral efficacy after day 6, possibly due to the development of drug-resistant strains.
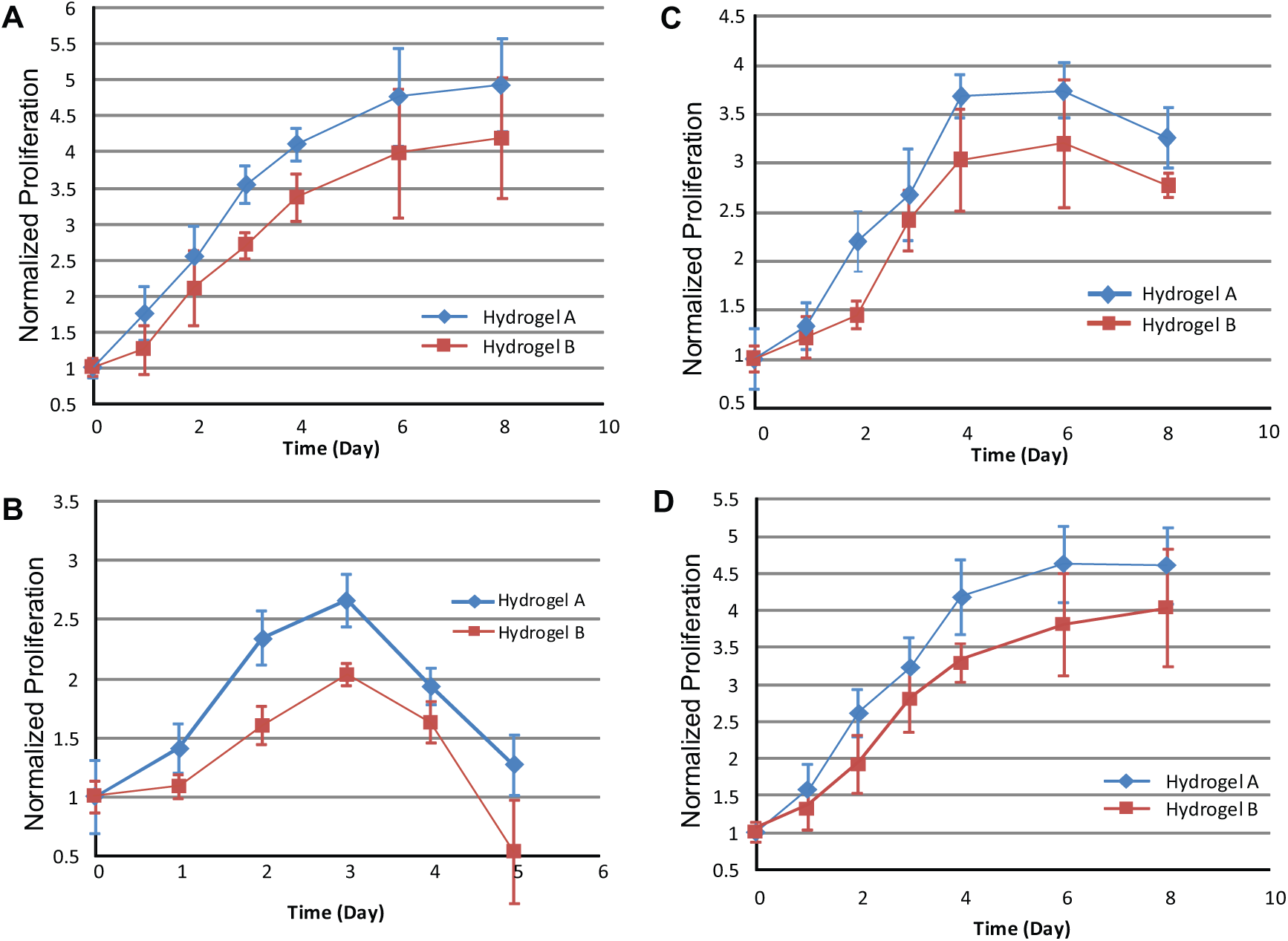
Proliferation of 3T3 fibroblasts in the modified HA hydrogel without any drug/virus treatments (
The hydrogel utilized in this culture model was designed to form covalently cross-linked hydrogels at 37 °C by using a tailorable recipe for tuning the composition proportions. The chemical cross-linking could proceed quickly in situ through a Michael-type reaction. The major advantage of this system is that the thermal gelation happens at physiological temperatures and favorable Michael addition cross-linking reaction occurs through mild processes and requires no exogenous, cytotoxic initiators, thus potentially allowing for efficacious examination of the impact of HSV-1 and ACV on cells.
The results indicate that ACV can efficiently block HSV-1 infection/replication in fibroblasts cultured in a 3D AHA hydrogel. In this study, HSV-1 and ACV could reach fibroblasts by a diffusion process through the AHA hydrogel network from the external culture medium. The virus could infect the cells encapsulated inside the 3D hydrogel, while ACV delivered an antiviral activity on the cells in 3D. However, high ACV concentrations also induce apoptosis in HSV-1-infected cells. For example, the ACV concentration should not be higher than 50 µg/mL in this hydrogel-based culture model, since a high drug concentration could cause more cell death after 2–4 days. The drug may take effect by potentiating some pathways that disrupt HSV-1 replication. 22 The 3D microenvironment could not directly influence the intracellular virus replication. Yet, the cure process for infected cells by ACV in this 3D culture model may also be influenced by the 3D AHA extracellular environment because 3D ECM may regulate the release of drug and the diffusion process of factors secreted by cells. This covalently cross-linked AHA hydrogel may serve as a culture model to mimic the antiviral activity of drugs delivered to the 3D ECM. 26 This AHA hydrogel–based in vitro model may be a candidate that allows us to screen for drugs that are highly effective against HSV-1 infection in a manner that is closer to the in vivo microenvironment. The study in the future may aim at further identifying the 3D ECM factors influencing the antiviral activity.
To summarize, the morphological evolution process of the multicellular structures presents obvious differences among control cells, cells treated with the virus alone, cells treated with the drug alone, and cells treated with both the virus and the drug. The cells treated with both the virus and the drug showed slower cellular growth and a subhealthy condition with less intercellular connections than the cells in the control sample, yet presented an overall higher metabolic activity after day 3 than the infected group. One interesting finding in this study is that cells were able to recover to normal spreading conditions and connect to each other after only 24 h in 2D culture, while the time for cells in 3D AHA gel to initiate this recovery process was significantly longer than that in traditional 2D culture. This hydrogel-based 3D culture model potentially lays the foundation for further studies on the impact of virus and drugs on the self-organized multicellular structures that mimic biological tissues.
Footnotes
Acknowledgements
The authors thank Prof. Tatiana Segura and Dr. Shiva Gojgini at University of California, Los Angeles for helpful discussion on the modified HA hydrogels. The authors also thank Wei Ma and Chunwang Xu for assisting in performing partial experiments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grants 51505127 and 81301293), the Natural Science Foundation of Jiangsu Province (grant BK20161197), the Changzhou Sci&Tech Program (grant CE20165029), the Major New Drugs Innovation and Development Project (2014ZX09507008), and Fundamental Research Funds for the Central Universities (grant 2015B04414) of China.