
Abstract
Tuberculosis still remains one of the most fatal infectious diseases. Streptomycin (SM) is the drug of choice, especially for patients with multidrug-resistant tuberculosis or category II patients, because it targets the protein synthesis machinery by interacting with steps of translation. Several mechanisms have been proposed to explain the resistance, but our knowledge is inadequate. Secretome often plays an important role in pathogenesis and is considered an attractive reservoir for the development of novel diagnostic markers and targets. In this study, we analyze the secretory proteins of streptomycin-resistant Mycobacterium tuberculosis isolates by 2-dimensional gel electrophoresis–matrix assisted laser desorption/ionization–time-of-flight mass spectrometry and bioinformatic tools. Fifteen overexpressed proteins were identified in a resistant isolate that belonged to various categories such as virulence/detoxification/adaptation, intermediary metabolism and respiration, and conserved hypotheticals. Among them, Rv1860, Rv1980c, Rv2140c, Rv1636, and Rv1926c were proteins of an undefined role. Molecular docking of these proteins with SM showed that it binds to their conserved domains and suggests that these might neutralize/compensate the effect of the drug. The interactome also suggests that overexpressed proteins along with their interactive partner might be involved in M. tuberculosis virulence and resistance. The cumulative effect of these overexpressed proteins could involve SM resistance, and these might be used as diagnostic markers or potential drug targets.
Introduction
Tuberculosis (TB) still remains one of the deadliest communicable diseases and was one of the top 10 causes of death worldwide in 2015. The World Health Organization (WHO) reported 10.4 million new TB cases and 1.8 million TB deaths, among them 0.4 million deaths resulting from TB among people living with human immunodeficiency virus (HIV). 1 The emergence of multidrug-resistant tuberculosis (MDR-TB) has worsened the situation, and streptomycin (SM) is still used to treat it. SM is the drug of choice, especially for patients with MDR-TB or category II patients. SM targets the translational machinery by interacting with several steps of translation. Several mechanisms have been proposed to explain SM resistance, but our knowledge is inadequate. The principal targets of SM resistance include mutation in the rpsL, rrs, and gidB genes (encoding the mutated product),2–4 enzymatic inactivation of drugs, 5 decreased inner membrane transport and active efflux pumps, 6 and cell wall impermeability. 7 SM resistance has been contributed by rpsL, rrs, and gidB gene mutation in approximately 60% to 70% of M. tuberculosis; however, the remaining 30% to 40% do not have these mutations, suggesting the involvement of some other mechanism(s). Advancement in expression proteomics has cleared the doubts of resistance phenotypes of those M. tuberculosis strains that do not have mutations in the genes responsible for resistance and might explore some other mechanism of resistance, such as enzymatic inactivation of antibiotic molecules, over expression of novel efflux pumps, and porin alterations in the cell wall, and trapping of drugs. As proteins manifest most of the biological processes, these are attractive targets for developing new drugs and diagnostics.8–13 Two-dimensional gel electrophoresis (2-DE) coupled with matrix assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF/MS) is the direct approach for separation, identification, and characterization of proteins and their species. 14 Comparative expression proteomic studies of aminoglycoside-resistant isolates have been reported.8–11 However, the secretory proteome of SM-resistant M. tuberculosis isolates has not been explored. To address this issue, we analyzed the secretory proteome of SM-resistant M. tuberculosis by direct proteomic and bioinformatics approaches. Such information could be helpful for the development of newer diagnostics and drug targets against SM resistance.
Materials and Methods
M. tuberculosis Isolates Collection and Drug Susceptibility Testing
Total susceptible (rifampicin, isoniazid, ethambutol, pyrazinamide, streptomycin, kanamycin, and amikacin) and SM-resistant (sensitive to other first-line and second-line drugs) M. tuberculosis isolates were procured from the Mycobacterial Repository Centre of the National JALMA Institute for Leprosy and Other Mycobacterial Diseases, Agra, India. Drugs susceptibility testing (DST) for all drugs was performed by the Löwenstein-Jensen (LJ) proportion 15 and resazurin microplate assay (REMA) methods. 16 Cultures were grown in Sauton’s liquid medium at 37 °C up to 4 weeks for proteomic analysis.
Secretory Protein Isolation and Precipitation
After 4 weeks, cells were removed by centrifugation; supernatant was sequentially filtered by 0.45- and 0.22-µm filters and precipitated using the sodium dodecyl sulfate (SDS)–trichloroacetic acid (TCA) precipitation method with the published protocol. 17 Protein concentrations were estimated by the Bradford method. 18 Protein extractions were performed in biological and technical replicas.
2D Gel Electrophoresis
Isoelectric focusing (IEF) and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were carried out using the published protocol of “in-gel rehydration” with slight modifications.8–11,19 Gel images were analyzed using PDQuest Advanced software version 8.0.0 (Bio-Rad, Hercules, CA). Protein spots that showed consistently increased intensities more than 1.5-fold were selected for identification. Student t test was used for the statistical analysis by PDQuest Advanced software. The system picks up the spots with differential intensity of significant levels built in the system. An equal amount of protein was loaded in all gels, and experiments were repeated in biological and technical replicates at least three times.
In-Gel Digestion and Mass Spectrometry
In-gel digestion of proteins and MALDI-TOF/MS were carried out using a published protocol.10,11,14,20 Mass spectra of digested proteins were acquired using the Autoflex II TOF/TOF 50 (Bruker Daltonik GmbH, Leipzig, Germany). Peptide mass tolerance was set to 50 to 125 ppm with carbamidomethyl-cysteine as the fixed modification. The oxidation of methionine was set as a variable modification, and one missed cleavage site was allowed.
Bioinformatic Analysis
Protein sequences of hypothetical proteins were retrieved from a Tuberculist server (http://tuberculist.epfl.ch). The probable conserved domain, interaction with drugs, and interacting partners were predicted using InterProScan, molecular docking, GPS-PUP,21-24 and STRING analysis (Search Tool for the Retrieval of Interacting Genes/Proteins; http://string.embl.de/).25,26 The STRING-10 server was used to predict the interacting partners of protein-protein interactions. The STRING database uses a combination of prediction approaches and an integration of other information (neighborhood, transferred neighborhood, gene fusion, co-occurrence, coexpression, experiments, databases, text mining). The network was made at a medium confidence level (0.400), allowing all active prediction methods.
Results
In this study, we have compared and analyzed the secretory protein profiles of SM-resistant and total sensitive isolates. Comparison of 2D gels ( Fig. 1 ) by PDQuest Advanced software revealed 16 protein spots that were consistently overexpressed in resistant compared with sensitive isolates (cut limit ≥1.5-fold changes in spot intensity). Student t test was used for statistical analysis with PDQuest software. Overexpressed proteins spots were identified by MALDI-TOF/MS and tabulated in Table 1 . The level of difference in protein spot intensity has been represented as a densitometric ratio in Table 1 .
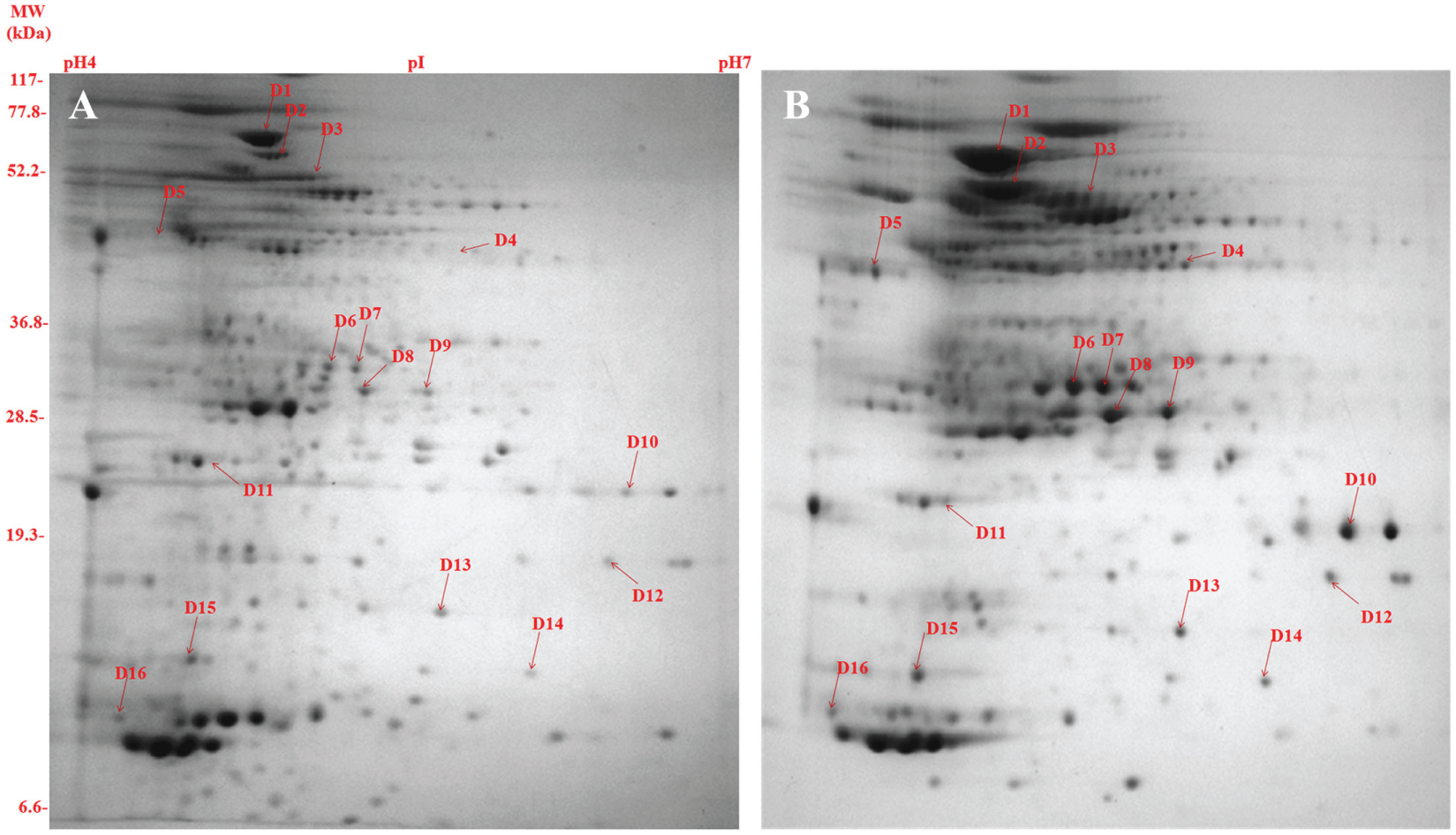
Composite images of the 2-dimensional gel electrophoresis profile of the secretory proteome of Mycobacterium tuberculosis clinical isolates: (
Details of Proteins Identified by Mass Spectrometry.
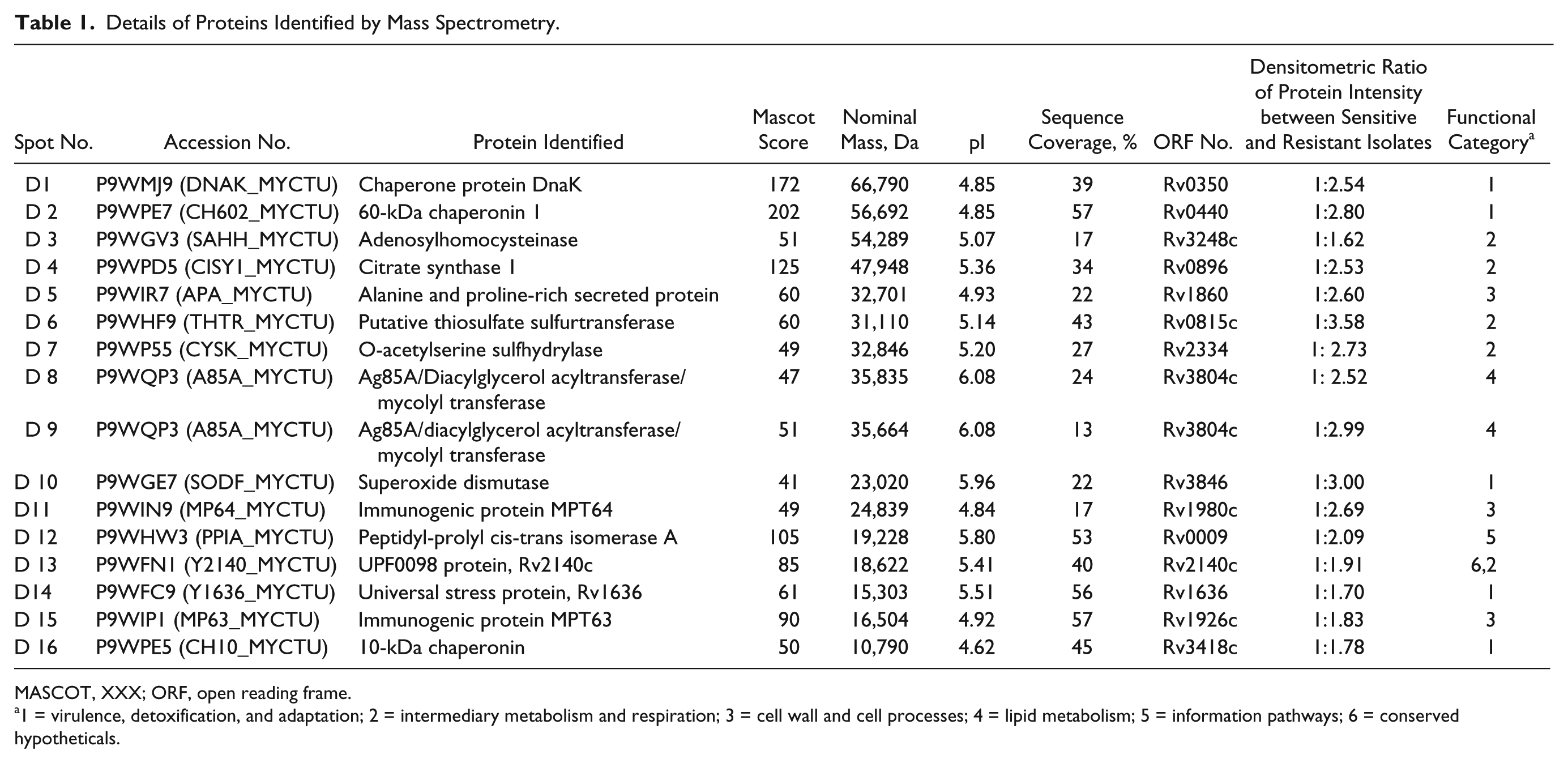
MASCOT, XXX; ORF, open reading frame.
1 = virulence, detoxification, and adaptation; 2 = intermediary metabolism and respiration; 3 = cell wall and cell processes; 4 = lipid metabolism; 5 = information pathways; 6 = conserved hypotheticals.
InterProScan Analysis
InterProScan analysis of proteins of undefined roles (Rv1860, Rv1636, Rv1980c, Rv2140c, and Rv1926c) showed the presence of their conserved domains ( Table 2 ).
InterProScan Analysis of Identified Proteins of Unknown Functions.
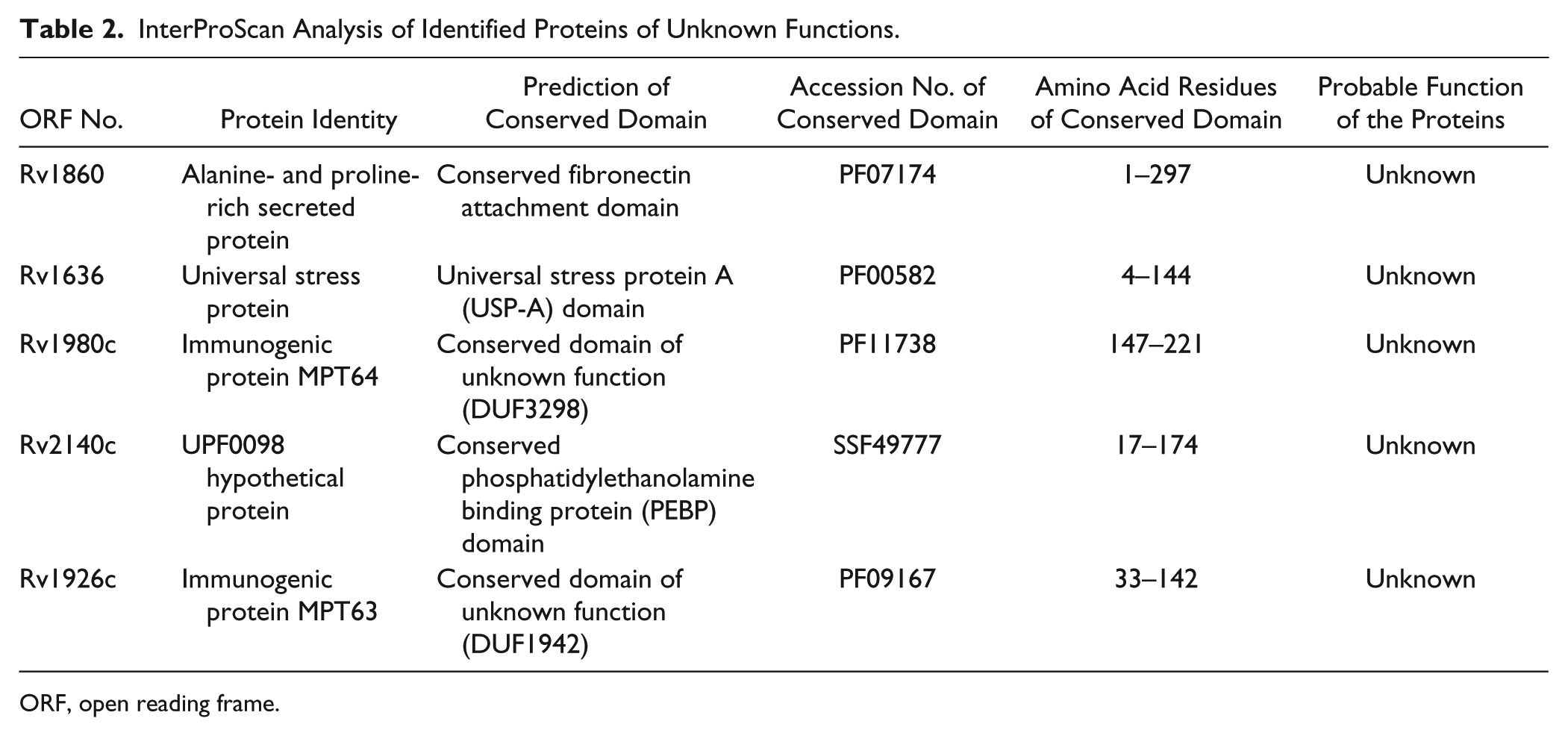
ORF, open reading frame.
3D Modeling and Docking
Molecular docking analysis of selected 3D models (showing less than 2% discrepancy from Ramachandran plot) of hypothetical proteins was performed to find their binding sites with SM. Parameters used for selection of 3D models and molecular docking are represented in Table 3 . Docking of Rv1860 ( Fig. 2A ) showed the interaction of SM with the amino acid residues of the conserved fibronectin attachment domain of alanine- and proline-rich secreted protein. Docking with Rv1636 ( Fig. 2B ) showed that SM bound to the residues of the universal stress protein A (USP-A) domain of the uncharacterized universal stress protein. Docking with Rv1980c ( Fig. 2C ) showed that SM bound to the interacting residues of the conserved domain of unknown function (DUF3298) of the immunogenic protein MPT64. Docking with Rv2140c ( Fig. 2D ) and Rv1926c ( Fig. 2E ) showed that SM interacted with amino acid residues of the conserved phosphatidylethanolamine binding protein (PEBP) domain of the hypothetical UPF0098 protein and domain of unknown function (DUF1942) of the immunogenic protein MPT63, respectively.
3D Modeling and Docking Parameters Used for Bioinformatic Analysis.
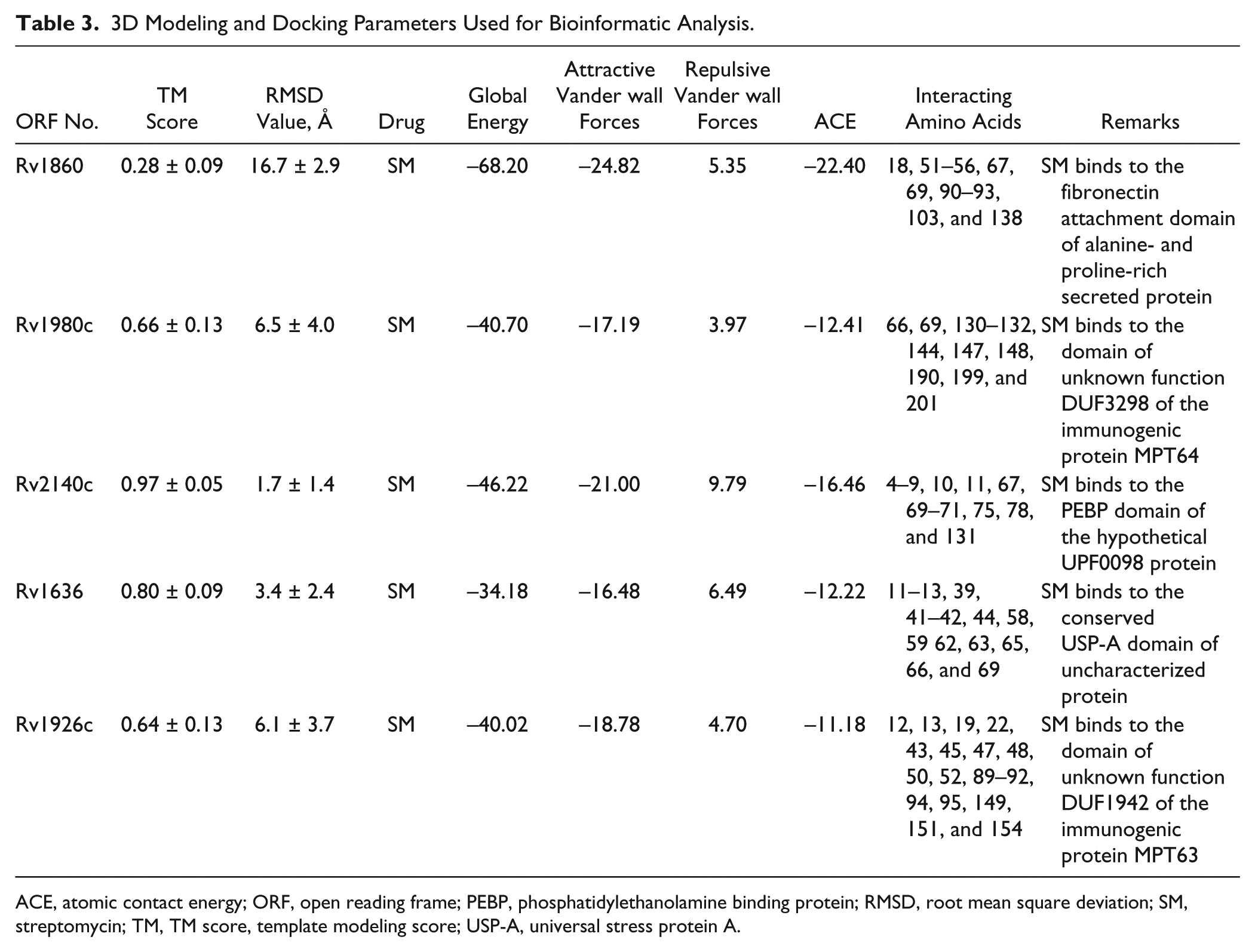
ACE, atomic contact energy; ORF, open reading frame; PEBP, phosphatidylethanolamine binding protein; RMSD, root mean square deviation; SM, streptomycin; TM, TM score, template modeling score; USP-A, universal stress protein A.
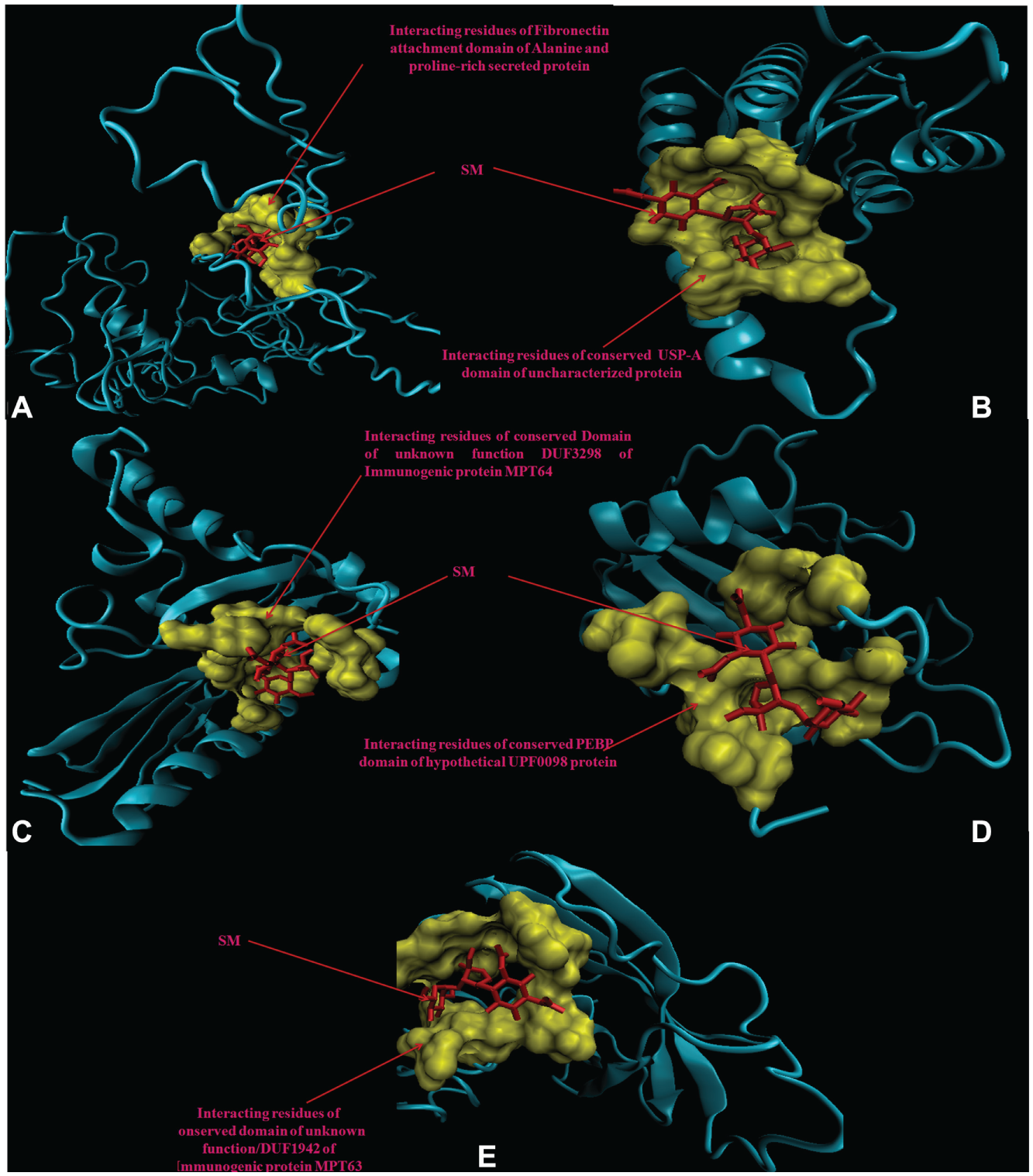
3D model of hypothetical proteins showing docking with streptomycin (SM) (red). (
Prediction of Pupylation Sites
Using the default threshold (medium) with a cutoff of 2.452, GPS-PUP predicted pupylation sites in 13 proteins, which were tabulated in Table 4 .
Predicted/Identified Pupylation Sites within Identified Proteins.
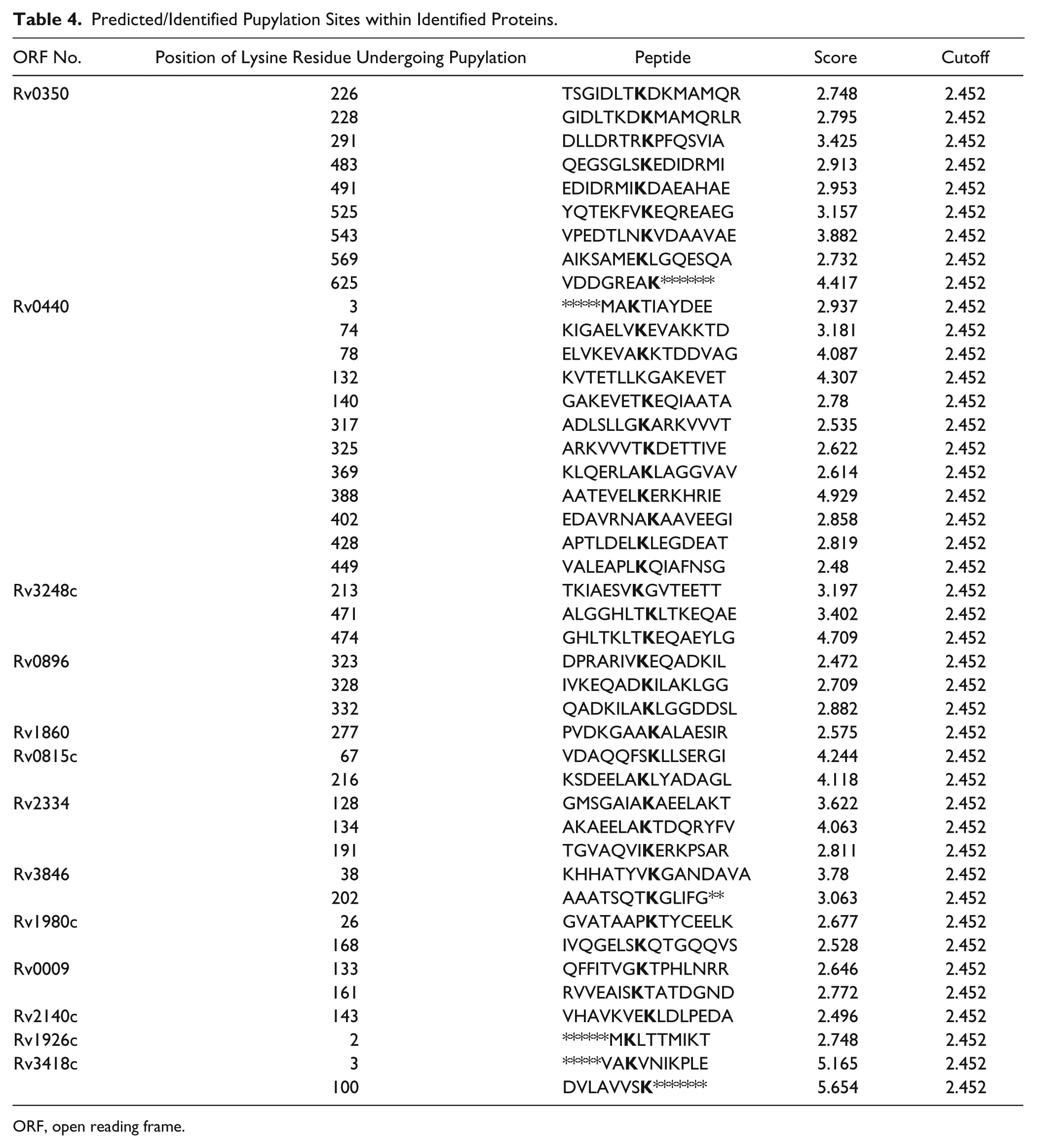
ORF, open reading frame.
STRING Analysis
We analyzed the overexpressed proteins using STRING-10 with a medium confidence score threshold of 0.4 and built an interactome network ( Fig. 3 ) for these proteins to find the protein-protein interaction (PPIs) and predict functional associations. We found that proteins involved in intermediary metabolism and respiration, lipid metabolism, information pathway/regulatory and virulence, and detoxification and adaptation interacted with each other as well as their partners, except the hypothetical proteins.
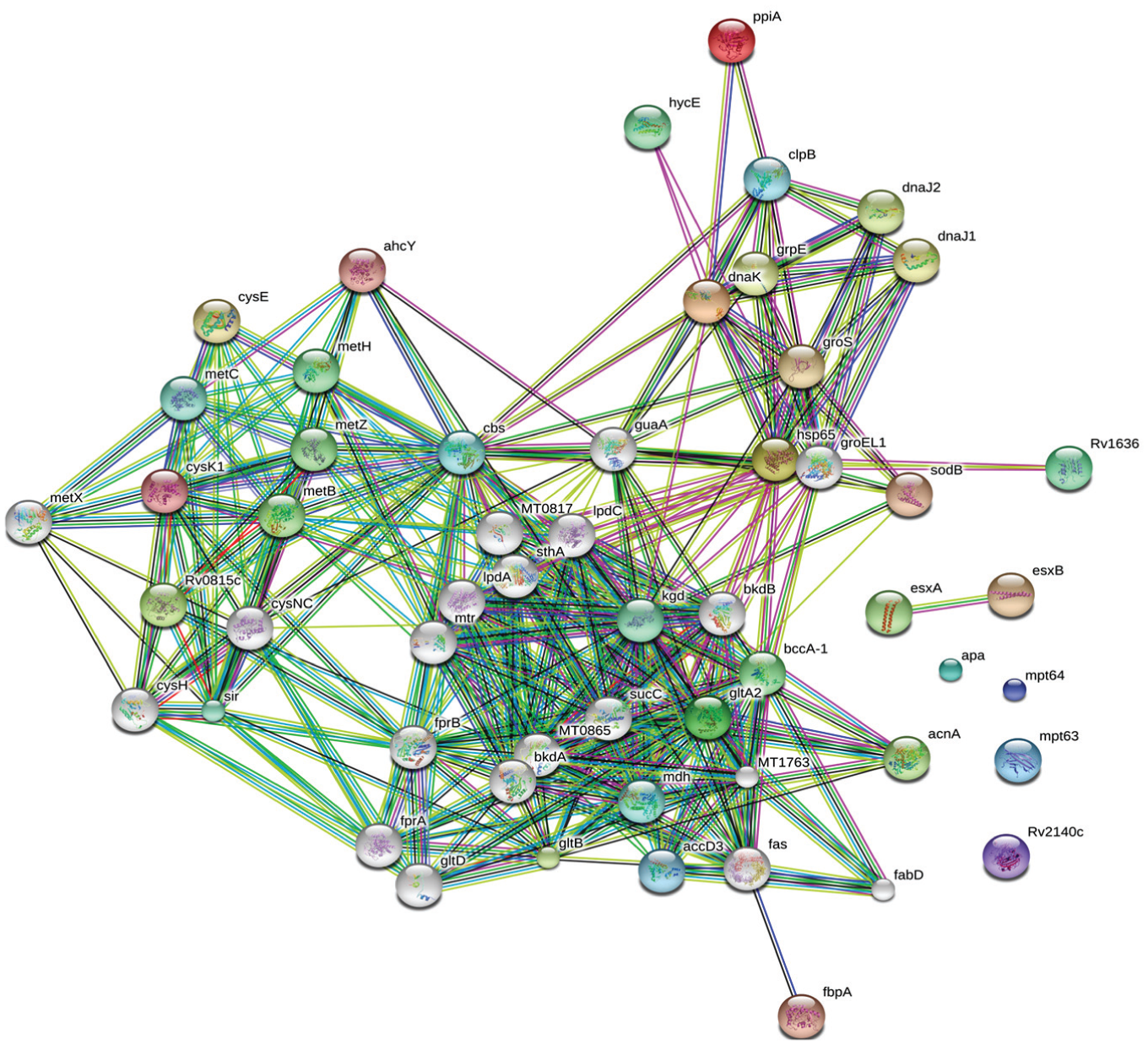
STRING analysis reveals the interaction partners of the overexpressed proteins that showed that overexpressed proteins interacted with proteins of the similar functional categories, except the uncharacterized proteins that had an undefined role.
Discussion
In this study, we have analyzed the secretory proteome of the SM-resistant isolate by 2DE-MALDI-TOF/MS and bioinformatic tools. Overexpressed proteins in the resistant clinical isolate were identified, which may be used as diagnostic markers or drug targets. 2DE/MS is the direct approach that is used for the identification of a large number of unknown proteins as well as for the separation of protein and its species, and it has an advantage over the traditional methods (SDS-PAGE, chromatography, and sequencing). Several reports on drug resistance and identification of diagnostics/drug targets employing proteomic approach exist.8–13,27 However, to our knowledge, no study on secretory proteome analysis with SM-resistant M. tuberculosis isolate has been reported.
Our study revealed 16 spots that were overexpressed in SM-resistant compared with total susceptible isolates. However, two spots were identified as one protein (isoforms of a protein or proteins of its species), and therefore a total of 15 proteins were found. Among them, Rv1860, Rv1980c, Rv2140c, Rv1636, and Rv1926c were proteins of an unknown function or undefined role. Of the 15, Rv0350, Rv0440, Rv3846, Rv1636, and Rv3418c belonged to virulence/detoxification/adaptation; Rv3248c, Rv0896, Rv0815c, and Rv2334 belonged to intermediary metabolism and respiration; Rv1860, Rv1980c, and Rv1926c belonged to cell wall and cell processes; and Rv3804c, Rv0009, and Rv2140c belonged to lipid metabolism, information pathways, and conserved hypothetical categories, respectively.
Rv0350 and Rv0440 are the 70-kDa and 60-kDa chaperone proteins respectively involved in refolding as well as proper assembly of unfolded/misfolded polypeptides under stresses. Rv0350 has a peptide binding domain as well as ATPase domain, which regulates the overexpression of other heat shock proteins. 28 Rv0440 is the second copy of 60-kDa chaperonin in the M. tuberculosis genome, which is situated far away from the classic GroEL-Cpn10 operon and suggests a specialized regulation in M. tuberculosis. 29 Sharma et al. 8 reported that these were overexpressed in SM-resistant M. tuberculosis. Overexpression of these might mitigate the toxicity by maintaining the proper folding of unfolded/misfolded protein or truncated proteins.
Rv3846, identified as superoxide dismutase, destroys free radicals that are normally produced within the cells under oxidative stress. Superoxide detoxifying enzyme, SodA (Rv3846), together with the thiol-oxidoreductase and an integral membrane protein, DoxX (Rv3005c), form a membrane-associated oxidoreductase complex (MRC). Loss of any MRC component is correlated with defective recycling of mycothiol, which is a functional analogue of glutathione in M. tuberculosis. 30 In the present study, overexpression of Rv3846 might protect M. tuberculosis cellular damage by mitigating the toxicity and lead to drug resistance.
Rv3804c has been identified as diacylglycerol acyltransferase/mycolyltransferase, which is involved in cell wall mycoloylation. It possesses a mycolyltransferase activity required for the biogenesis of trehalose dimycolate (cord factor) and maintaining cell wall integrity, which leads to persistent tuberculosis infection. 31 In our study, overexpression of Rv3804c might be protect the M. tuberculosis to persist leads to development of streptomycin resistance.
Rv0815c is the putative thiosulfate sulfurtransferase (Rhodanese-like protein) involved in the formation of thiosulfate and probably protects the becillai from oxidative stress. Singhal et al. 32 reported that Rv0815c was overexpressed intraphagosomally in THP-1 cells infected by a drug-resistant strain of M. tuberculosis. Overexpression of Rv0815c might be protect the M. tuberculosis from streptomycin (SM) drug resistance.
Chaperonin/10-kDa antigen (a subgroup of molecular chaperones) has homology with the GroES (Rv3418c) or chaperonin-10/family of heat shock proteins. Kumar et al. 9 reported the overexpression of Rv3418c in amikacin- and kanamycin-resistant M. tuberculosis. Recently, Lata et al. 33 reported the higher expression of Rv3418c in ofloxacin- and moxifloxacin-induced M. tuberculosis. Overexpression of this protein might mitigate the toxicity by maintaining the proper folding of unfolded/misfolded protein or truncated proteins.
We hypothesized that overexpression of Rv0350, Rv0440, Rv3846, Rv3804c, Rv0815c, and Rv3418c proteins might cumulatively mitigate the toxicity and contribute to SM resistance.
Rv3248c is adenosylhomocysteinase that catalyzes the reversible hydrolysis of S-adenosylhomocysteine (SAH) into free adenosine and L-homocysteine. SAH is produced from the by-product of a methylation reaction catalyzed by S-adenosyl methionine (SAM)–dependent methyltransferase. Methylation involved in various cellular and metabolic biosynthesis pathways and adenosylhomocysteinase is considered “druggable.” 34 SM-resistant mycobacteria also are dormant due to repression of targets, which is the mycobacterial intrinsic defense mechanism against resistance. Overexpression of Rv3248c might maintain a supply of precursors of cellular and metabolic biosynthesis pathways to resistant M. tuberculosis.
Rv0896 (citrate synthase 1) is involved in the tricarboxylic acid cycle that is the central metabolic pathway and responsible for the synthesis of many important precursors and molecules. 35 Overexpression of Rv0896 might maintain an energy supply to resistant M. tuberculosis.
Rv2334 (O-acetylserine sulfhydrylase) catalyzes the conversion of O-acetylserine (OAS) to cysteine, which is the precursor of the amino acid biosynthesis pathway. Our study assumes that overexpression of this protein regulates the supply of amino acids for translation steps of mycobacteria even in the dormant state.
Rv0009 (probable iron-regulated peptidyl prolyl cis-trans isomerase A) is probably involved in cis-trans isomerization of proline imidic peptide bonds in oligopeptides and accelerated protein folding. Lata et al. 33 reported the higher expression of Rv0009 in ofloxacin- and moxifloxacin-induced M. tuberculosis. Recently, Sharma et al. 11 reported the higher expression of Rv0009 in aminoglycoside-resistant M. tuberculosis. Overexpression of Rv0009 might maintain an amino acid supply to cells even in a low translational state or dormant state.
We hypothesized that overexpression of Rv3248c, Rv0896, Rv2334, and Rv0009 proteins might cumulatively overcome the effect of targets repression and contribute to SM resistance.
Rv1636, identified as a universal stress protein with unknown function, is overexpressed in nutrient/oxygen-deficient conditions and is involved in virulence/chronic infection. 36 Lata et al. 33 reported that Rv1636 was overexpressed in ofloxacin- and moxifloxacin-induced M. tuberculosis. Recently, Sharma et al. 11 reported that it was overexpressed in aminoglycoside-resistant isolates. In the present study, Rv1636 was overexpressed, and molecular docking predicted that SM binds to the residues of conserved USP-A domains, which might be altering the functions. It is suggested that overexpression of Rv1636 might neutralize/compensate the effect of the drug.
Rv2140c (UPF0098 protein) is the uncharacterized protein of unknown function that had a PEBP domain. 37 Lata et al. 27 reported that Rv2140c was overexpressed in ofloxacin-resistant M. tuberculosis. Recently, Sharma et al. 11 reported that Rv2140c was overexpressed in aminoglycoside-resistant M. tuberculosis. In the present study, we have observed that SM binds to the residues of the conserved PEBP domain, which might be altering the functions.
Rv1860 is identified as alanine- and proline-rich secreted protein Apa (uncharacterized), which has the fibronectin attachment domain. Recently, Singhal et al. 38 reported that the Mycobacterium bovis BCG alanine- and proline-rich secreted protein was intraphagosomally overexpressed in THP-1 cells. In this study, we predicted that SM binds to residues of the conserved fibronectin attachment domain of Rv1860 and suggested that overexpression of this protein might neutralize/compensate the effect of the drug.
Rv1980c and Rv1926c are the uncharacterized immunogenic protein MPT64 and immunogenic protein MPT63 that have domain of unknown function DUF3298 and DUF1942, respectably. Recently, Singhal et al. 38 reported that the M. bovis BCG alanine- and proline-rich secreted protein was intraphagosomally overexpressed in THP-1 cells. In this study, we predicted that SM binds to residues of conserved domains of unknown function and suggested that overexpression of this protein might neutralize/compensate the effect of the drug.
We hypothesized that overexpression of Rv1636, Rv2140c, Rv1860, Rv1980c, and Rv1926c uncharacterized/hypothetical proteins might cumulatively neutralize/compensate the effect of drugs and contribute to SM resistance.
Pupylation is a reversible posttranslational modification (PTM) that is likely to have a regulatory role. 39 Pupylation contributes to the virulence/survival strategy of M. tuberculosis in the host and makes the bacteria more resistant to drug and other stresses.10,11,40 In our study, 13 overexpressed proteins showed the pupylation sites, which suggested that overexpression of these proteins via the Pup-proteasome system (protein-protein interaction) might be involved in protein turnover to overcome SM stress.
STRING analysis revealed that overexpressed proteins of virulence, detoxification, and adaptation (VDA) category, intermediary metabolism and respiration (IMR), lipid metabolism (LM), information pathway (IP) and interacted to the other proteins which were involved in pathways of virulence, detoxification, and adaptation (VDA), intermediary metabolism and respiration (IMR), lipid metabolism (LM), and IP (information pathways) categories. We suggested that overexpressed proteins along with their interactive partners might cumulatively be involved in SM resistance. We assume that overexpressed proteins might be contributing to SM resistance and acting as a diagnostic marker or potential targets for drug development against resistance.
In conclusion, in this report, we focus on the secretory proteome of SM-resistant M. tuberculosis using a proteomic and bioinformatic approach. Among the 15 overexpressed proteins, Rv1860, Rv1980c, Rv2140c, Rv1636, and Rv1926c were proteins of unknown function or undefined role. Molecular docking of these five uncharacterized proteins (Rv1860, Rv1980c, Rv2140c, Rv1636, and Rv1926c) showed that SM interacted with their conserved domains. GPS-PUP predicted the presence of pupylation sites within 13 proteins. The interactome also suggested that overexpressed proteins interacted with each other and their partners except the hypothetical proteins. It is depicted that overexpression of these proteins might not only neutralize/modulate the drug molecules but also be involved in various mechanisms such as mitigating the toxicity and repressing drug target and protein turnover to overcome SM resistance. We assume that the cumulative effect of these overexpressed proteins might be responsible for SM resistance. These findings need further exploitation for the development of newer drug entities or diagnostic markers.
Footnotes
Acknowledgements
We thank Director, National JALMA Institute for Leprosy and Other Mycobacterial Diseases for support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a postdoctoral research fellowship grant from the Indian Council of Medical Research (No. 3/1/3/PDF(11)/2016/-HRD), New Delhi, India.