
Abstract
Deletion of a single phenylalanine residue at position 508 of the protein CFTR (cystic fibrosis transmembrane conductance regulator), a chloride channel in lung epithelium, is the most common cause for cystic fibrosis. As a consequence, folding of the CFTRΔF508 protein and delivery to the cell surface are compromised, resulting in degradation of the polypeptide. Accordingly, decreased surface presence of CFTRΔF508 causes impaired chloride ion conductivity and is associated with mucus accumulation, a hallmark of cystic fibrosis. Molecular chaperones such as Hsp90 and its co-chaperone partner Aha1 are thought to play a key role in targeting folding-deficient CFTRΔF508 for degradation. Thus, pharmacologic manipulation to inhibit Hsp90-Aha1 chaperone complex formation appears beneficial to inhibit proteolysis of CFTRΔF508 and rescue its residual chloride channel activity. Therefore, we have screened a collection of 14,400 druglike chemical compounds for inhibitors of the Hsp90-Aha1 complex by amplified luminescence proximity homogeneous assay (Alpha). We identified two druglike molecules that showed promising results when we tested their ability to restore chloride channel activity in culture cells expressing the mutant CFTRΔF508 protein. The two molecules were most effective in combination with the corrector VX-809 and may therefore serve as a lead compound that can be further developed into a drug to treat cystic fibrosis patients.
Keywords
Introduction
One out of 2000 to 3000 newborns in the white Caucasian population is affected by cystic fibrosis (CF), also known as mucoviscidosis, an inherited recessive disorder. Hallmarks of the malady are progressive lung disease, the most common cause for mortality; pancreatic dysfunctions; intestinal obstruction; and male infertility. Cystic fibrosis is characterized by accumulation of viscous mucus in the epithelia including the airways, leading to dysfunction and bacterial infections. Although several hundred mutations are known, the deletion of a phenylalanine residue at position 508 of the polypeptide cystic fibrosis transmembrane conductance regulator (CFTR) accounts for about 70% of all cases. CFTR, a chloride channel, mediates ion conductance across cellular membranes. Mutant CFTR that lacks phenylalanine 508 (CFTRΔ508) is incorrectly processed, fails to be delivered from the endoplasmic reticulum to the plasma membrane, and is associated with defective osmotic water transport and mucus accumulation. 1 It is believed that the mutation leads to misfolding of the CFTRΔ508 protein, which is recognized by the cellular protein quality control and disposed by the proteosomal degradation machinery. Folding and quality control of CFTRΔ508 involve cytosolic chaperones such as the Hsp70 and Hsp90 systems and CHIP, an Hsp70/Hsp90 co-chaperone with ubiquitin ligase activity ( Fig. 1 ).2,3 Recent studies have presented evidence that the Hsp90 co-chaperone and Hsp90ATPase activator Aha14,5 may play a role in triaging CFTRΔ508 for degradation.6,7 It was reported that the Hsp90-Aha1 complex associates preferentially with the folding deficient CFTRΔF508 protein through specific interaction mediated by the co-chaperone Aha1, leading to subsequent protein quality control and degradation. In these studies, the Hsp90α isoform was identified as a direct CFTRΔF508 binding partner by mass spectrometry. 7 Therefore, uncoupling Aha1 from Hsp90 may rescue the chloride channel from proteolytic breakdown. 8 In this study, we have adapted the Hsp90-Aha1 protein complex to serve as a target for inhibitor screening by Alpha technology. Hit compounds were tested for their potency to restore CFTRΔF508-associated membrane conductivity in culture cells. We have identified two compounds that increased ion efflux ~2.5-fold and had even a potentiated synergistic effect when administered together with the corrector VX-809.
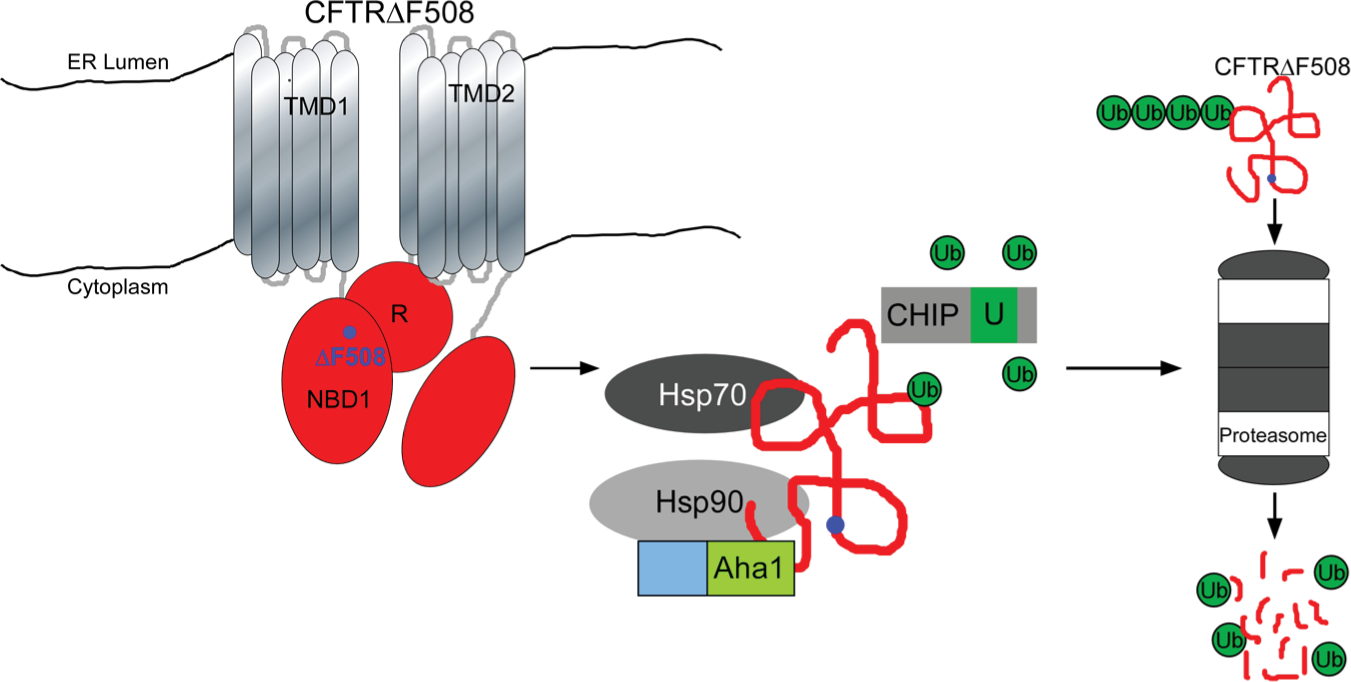
The Hsp90-Aha1 chaperone complex participates in protein quality control of mutant CFTRΔF508 protein. Hsp90 and Aha1 together with the molecular chaperone Hsp70 and the E3 ubiquitin ligase CHIP triage CFTRF508 for ubiquitination and subsequent degradation by the proteasome system. TMD = transmembrane domain; NBD = nucleotide binding domain.
Materials and Methods
Analysis of Hsp90-Aha1 Complex Formation by Gel Filtration Chromatography
Complex formation of GST-Aha1 and His6-Hsp90 was analyzed by gel filtration chromatography using purified recombinant proteins as described. 5 Briefly, both proteins were mixed in 40 mM HEPES-KOH buffer pH 7.4, 50 mM KCl, and 2 mM MgCl2 and separated on a Superdex 200 HR 10/30 column using an ÄktaPurifier system. Fractions were collected and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 10% gels.
High-Throughput Screening for Inhibitors of the Hsp90-Aha1 Complex by Alpha Assay
GST-tagged human Aha1 (GST-Aha1) and His6-tagged human Hsp90α (His6-Hsp90) were expressed in Escherichia coli and purified by affinity chromatography on glutathione and on nickel-nitriloacetate chelate affinity resins, respectively, followed by ion exchange chromatography using a ResourceQ column operated by an Äkta purifier system (GE Healthcare). Five microliters of GST-Aha1 (1.5 µM) and His6-Hsp90 (1.5 µM) was mixed to form complexes and incubated in 40 mM HEPES pH 7.4, 50 mM KCl, 2 mM MgCl2 with 5 µl of a druglike compound (50 µM) from the Maybridge HitFinder collection. The mixtures were incubated for 10 min at 30 °C followed by 10 min at 4 °C. Finally, 10 µL of donor and acceptor beads in 40 mM HEPES pH 7.4, 50 mM KCl, 2 mM MgCl2, 0.5% bovine serum albumin were added at a final concentration of 4 µg and further incubated at room temperature for 40 min in an AlphaPlate-384 (PerkinElmer, Waltham, MA). Accordingly, the Hsp90-Aha1 complex was at a final concentration of 0.3 µM with the potential inhibitor at 10 µM. Control reactions received 1 M NaCl to destroy the Hsp90-Aha1 complex. To analyze the robustness of the assay, we measured the Alpha signals of 48 wells containing the Hsp90-Aha1 complex and of another 48 wells containing the negative control reactions. The Z′ factor of the assay was determined according to the equation Z′ = 1 – (3*SDhigh + 3*SDlow)/(Meanhigh – Meanlow) as described before. 9
To test all 14,400 compounds of the Maybridge HitFinder collection, we used a Liquidator 96 pipetting system and performed assays in a 384-well plate format. Plates were counted in an EnSpire Multimode Plate Reader (PerkinElmer). For counterscreening, a GST-His6 peptide was expressed and purified from E. coli and used as a constitutive linker between donor and acceptor beads. Positive hits were further confirmed in triplicate. To determine IC50 values, 0.0001, 0.001, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30, and 100 µM of the compounds were incubated with 0.3 µM of the Hsp90-Aha1 complex and measured by Alpha assay as described above. The experiments were done in triplicate, and data were fitted by a four-parameter logistic curve equation using SigmaPlot software.
Iodide Efflux Assay to Test Hit Compounds as Candidate Drugs for Their Potency to Preserve Residual Channel Activity of the CFTRΔF508 Protein
We used BHK cells stably expressing either wild-type CFTR or the CFTRΔF508 mutant, a kind gift from Dr. Margarida Amaral, to test our candidate drugs in a functional assay. Cells were cultured in Ham’s F12 medium supplemented with 5% fetal calf serum and 500 µM methotrexate at 37 °C. Twenty-four hours after plating, cells were treated with candidate drugs or the corrector VX-809 as a positive control. For the iodide efflux assay, cells were bathed in loading buffer (136 mM NaI, 3 mM KNO3, 2 mM Ca[NO3]2, 20 mM HEPES pH 7.4, 11 mM glucose) and then washed 10 times with efflux buffer (136 mM NaNO3, 3 mM KNO3, 2 mM Ca[NO3]2, 20 mM HEPES pH 7.4, 11 mM glucose) to remove NaI from the medium. Efflux of iodide from cells was induced by addition of 10 µM forskolin and 50 µM genistein to stimulate the CFTR channel, and the release of the ion was measured every minute for 10 min using an iodide-sensitive electrode.7,10 Total iodide efflux was calculated from the up to 10 measured values of this time course that were above baseline, and the experiment was done in triplicate.
Results and Discussion
We have reconstituted the complex between GST-Aha1 and His6-Hsp90 using purified proteins as shown by gel filtration chromatography ( Fig. 2A ). Subsequently, the proteins were attached via their GST-tag to GSH (glutathione) coupled donor beads and via their His6-tag to nickel chelate coupled acceptor beads to form an Aha1-Hsp90 complex that serves as a target for Alpha screening ( Fig. 2B ). Donor and acceptor beads are brought into close proximity by the Aha1-Hsp90 interaction. Upon laser excitation, donor beads produce singlet oxygen (1O2) that diffuses to the acceptor bead and triggers emission of luminescent light. Molecules that inhibit the protein-protein interaction keep donor and acceptor beads apart and suppress luminescence emission by acceptor beads. The decrease in luminescence serves as a readout to quantify the inhibitory effect of a chemical hit compound on the protein interaction. For counterscreening, donor and acceptor beads were connected by a GST-His6 fusion peptide. True hits do not interfere with light emission in this setup.
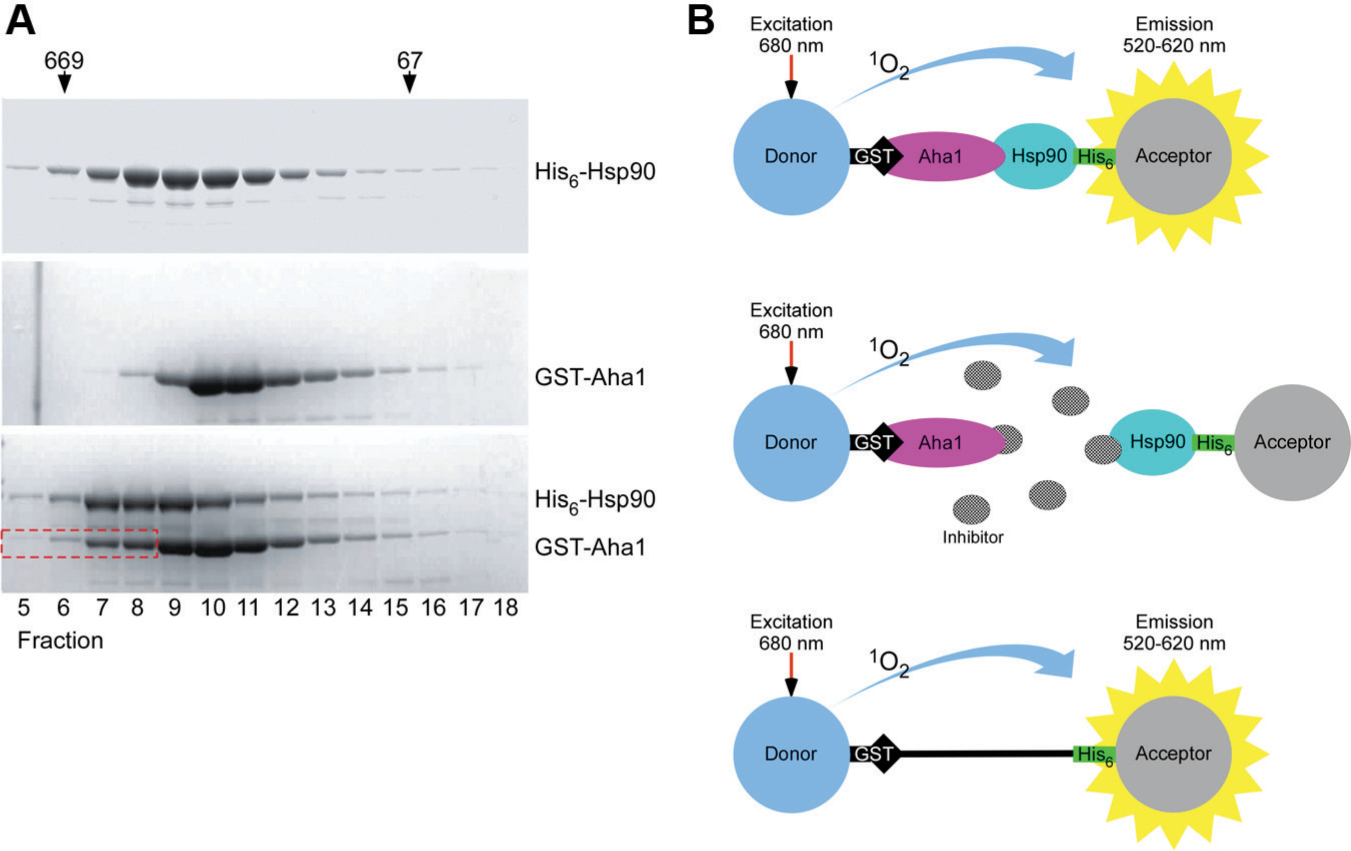
Principle of the Alpha screen technology using the Hsp90-Aha1 protein complex. (
To check the robustness of the screening procedure, we measured 48 wells containing the Hsp90-Aha1 complex and 48 wells containing the negative control reaction. The average signal for the Hsp90-Aha1 complex (Meanhigh) was 40,883 with a standard deviation (SDhigh) of 2314. The average signal for the negative control was (Meanlow) 286 with a standard deviation (SDlow) of 228. The Z′ factor calculated from these data was 0.81 according to the formula described in the Materials and Methods section, suggesting that the robustness of the assay is excellent. Next, the 14,400 druglike molecules of the Maybridge HitFinder collection were tested at 10 µM for their ability to inhibit the Hsp90-Aha1 interaction. In the primary screen, we identified 30 candidate druglike molecules out of 14,400 that inhibited Hsp90-Aha1 complex formation ≥90% (hit rate 0.2%). Eleven molecules passed rescreening and the counterscreening procedure. Three compounds were toxic to BHK cells in culture and therefore were not used for further experiments. Slow-folding proteins such as the Hsp90 client CFTRΔF508 benefit from a decelerated adenosine triphosphate (ATP) hydrolysis rate of the molecular chaperone. Accordingly, disturbing the interaction with the ATPase stimulator Aha1 may slow down the ATP-driven conformational cycle of Hsp90, increase the dwell time with CFTRΔF508, and therefore enhance folding efficiency of this slow-folding client protein. 11 Therefore, the remaining eight candidate molecules from our screening procedure ( Fig. 3A ) were tested for their potency to promote ion release from BHK cells stably expressing mutant CFTRΔF508 protein by iodide efflux assay. Treatment with 5 µM of compounds A12 and A16 induced iodide release from BHK CFTR F508 cells ~2.5-fold compared with the untreated control, suggesting that the two druglike molecules correct the mutant CFTRΔF508 protein and preserve its residual chloride channel activity ( Fig. 3B ). The other six compounds showed no effect or even decreased iodide efflux ( Fig. 3B ). The IC50 values for A12 and A16 were determined at an Hsp90-Aha1 complex concentration of 0.3 µM and are indicated in Figure 4A . Next, we examined whether A12 and A16 may act together with the corrector VX-809. It is thought that VX-809 directly binds to the interface of CFTR’s NBD1 with neighboring segments and stabilizes CFTRΔF508 that has made its way to the cell surface to prevent further protein steps of protein quality control. 12 When A12 and A16 were combined with VX-809, a potentiated synergistic effect was observed that resulted in an ~25-fold or 15-fold release of iodide compared with the control, respectively ( Fig. 4B , C ). This finding proposes strategies to use combinations of multiple drugs to treat CF and suggests that A12 and A16 act within the same molecular pathway together with VX-809. This result fits other studies that have reported compounds that increase VX-809-mediated CFTRΔF508 activity. 13 VX-809, together with A12, A16, and other correctors, appears to affect different steps of a common quality control pathway necessary to prevent degradation of mutant CFTRΔF508 and restore its chloride channel activity.14,15 To further prove the effect of A12 and A16, one may consider using primary airway cells from ΔF508 CF patients as a more physiological model. Hsp90-Aha1 complex inhibitors that decrease iodide efflux, such as A10 (see Fig. 3 ), may interact with additional proteins that increase degradation or inhibit activity of CFTRΔF508. 16 The molecules identified to inhibit Hsp90-Aha1 complex formation as shown in Figure 3A do not share a common structural motif. Therefore, they may bind either Hsp90 or Aha1 or at different places of the wide-stretched interaction site between the two binding partners. 17
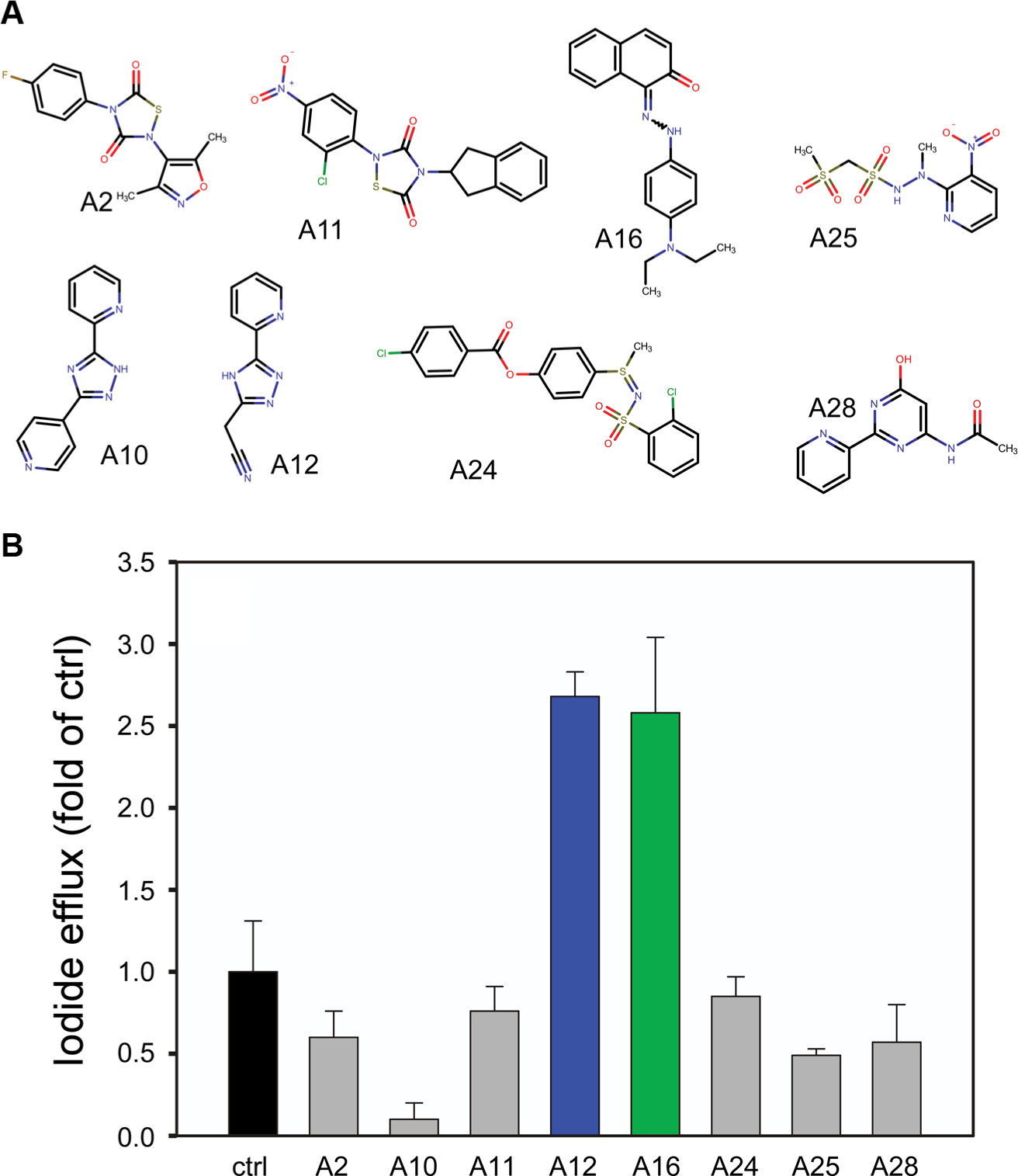
Identification of Hsp90-Aha1 complex inhibitors by Alpha screening. (
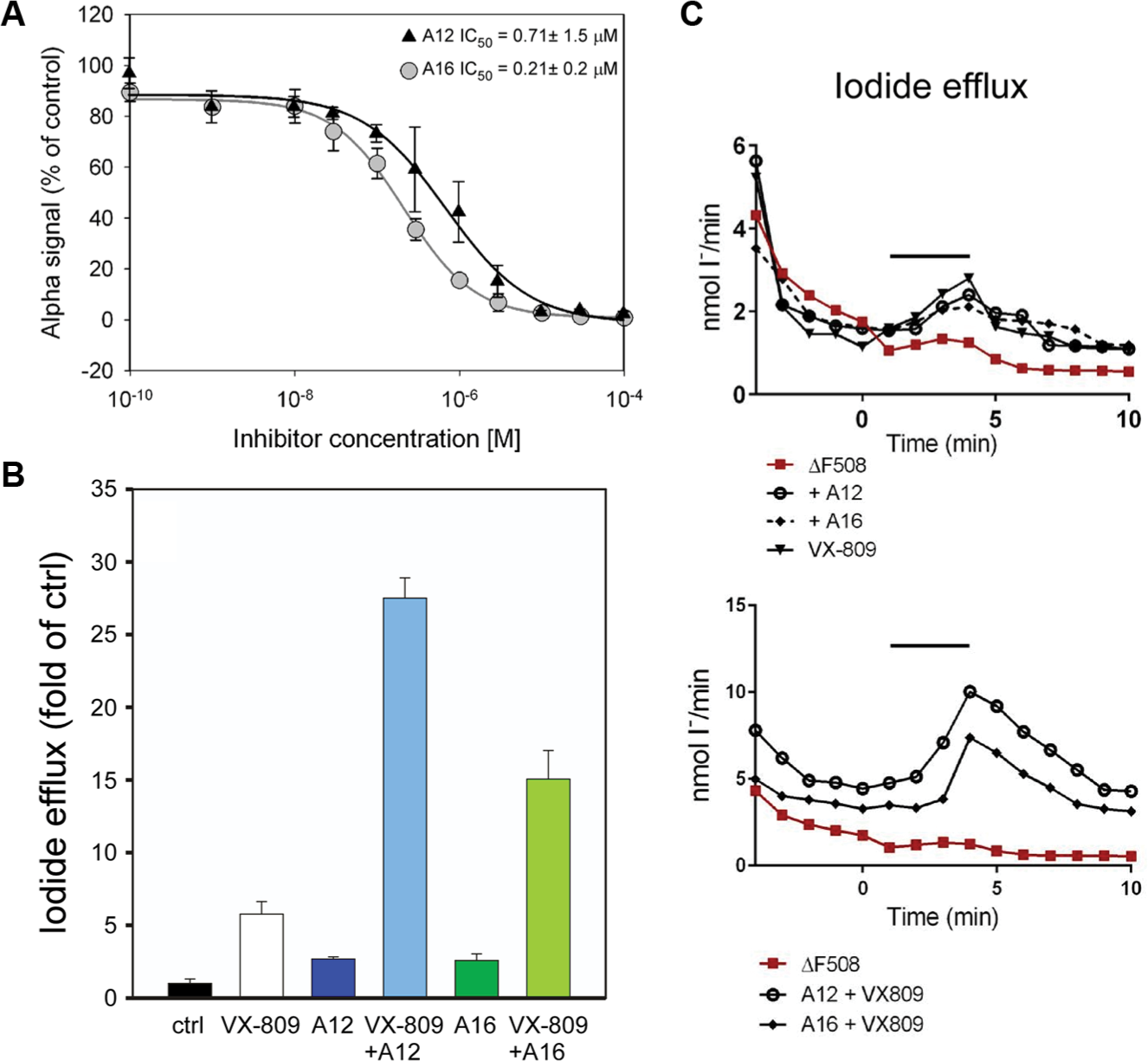
Compounds A12 and A16 inhibit Hsp90-Aha1 complex formation and raise iodide efflux synergistically together with the corrector VX-809. (
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by financial grant S03/10 from Mukoviszidose Institut gGmbH, Bonn, Germany, the research and development arm of the German Cystic Fibrosis Association Mukoviszidose e.V. (to W.M.J.O.) and in part by Ruhr-University Bochum FoRUM grant F829 (to W.M.J.O.).