
Abstract
Lipopolysaccharide (LPS) is the main structural component of the outer membrane of most Gram-negative bacteria and has diverse immunostimulatory and procoagulant effects. Even though LPS is well described for its role in the pathology of sepsis, considerable evidence demonstrates that LPS-induced signalling and immune dysregulation are also relevant in the pathophysiology of many diseases, characteristically where endotoxaemia is less severe. These diseases are typically chronic and progressive in nature and span broad classifications, including neurodegenerative, metabolic, and cardiovascular diseases. This Review reappraises the mechanisms of LPS-induced signalling and emphasises the crucial contribution of LPS to the pathology of multiple chronic diseases, beyond conventional sepsis. This perspective asserts that new ways of approaching chronic diseases by targeting LPS-driven pathways may be of therapeutic benefit in a wide range of chronic inflammatory conditions.
Introduction
Considerable research into the impact of the microbiome on human health strongly implicates a role for microorganisms, their molecular products, and the host defences against them in the complex aetiopathogenesis of a broad range of chronic inflammatory diseases.1,2 Various bacterial components are recognised by the innate immune system. One of the most well studied of these is lipopolysaccharide (LPS; also known as endotoxin) that derives from the cell wall of Gram-negative bacteria such as Escherichia coli, Salmonella species, Porphyromonas species and Helicobacter species. Once in the circulation (known as endotoxaemia) LPS has a marked stimulatory effect on the immune system, and LPS-mediated signalling can cause a range of pathology. 3 In severe cases this can lead to sepsis. 4
Although much of the research into the physiological effects of LPS has been carried out in the setting of sepsis, LPS signalling is highly relevant to the pathophysiology of many chronic inflammatory diseases, including neurodegenerative disease, metabolic disease and cardiovascular disease, and particularly at LPS levels that sustain low-grade non-resolving inflammation. 3 LPS should therefore be regarded in chronic diseases, even if it is present in the body at only low concentrations.
In light of these newly appreciated disease associations, we reappraise the roles of LPS signalling in the pathology of chronic diseases. We consider the structural aspects of LPS that are relevant to the immune response and how LPS, together with its effectors, influence immune signalling. We discuss the consequence of LPS-induced signalling in chronic inflammatory diseases and summarise the therapeutic knowledge generated from sepsis research that might be repositioned to manipulate LPS-driven pathways in a broad range of chronic, inflammatory diseases.
LPS: a Bacterial Wall Component
LPS is the main component of the outer membrane of most Gram-negative bacteria. LPS is composed of a lipid A hydrophobic anchor, a proximal non-repeating oligosaccharide core, and a distal chain that is typically attached to a polymer of repeating saccharide subunits called the O-antigen that extends further from the bacterial surface (eg5,6) (Figure 1a). LPS is a potent stimulator of innate immunity in the host, with the lipid A moiety having most of the immunostimulatory activity (eg7,8). Structural variation of lipid A can alter the stimulatory capacity of LPS, which is related to patterns of acylation and phosphorylation and the presence of charged groups. 8 Diversity in the polysaccharide chain also modulates the potency of LPS, and defines the serotype of the bacterium. 6

The structures of LPS and LBP. (a) Simplified structure of LPS from Gram-negative bacteria. The immunostimulatory lipid A is bound to a conserved inner core of sugars such as Kdo, which is bound in turn to a more variable outer core of common hexose sugars (eg glucose and galactose). The highly diverse O-antigen at the distal region of the LPS molecule is composed of repeated units of common hexose sugars. Rough-type LPS lacks an O-antigen. (b) Crystal structure of LBP (Protein Data Bank: 4M4D). The N-terminal portion is the main binding sites for lipid A, with the C-terminal involved in LPS transfer.
Once LPS is synthesised and exported to the bacterial outer membrane, small amounts of LPS (membrane fragments) may be liberated from the bacterial surface during replication; larger amounts are set free during death or lysis. LPS is also secreted from bacteria through the production of outer membrane vesicles (OMVs). 9 As LPS is an amphiphilic molecule, it can form micelles (supramolecular aggregates) above a critical concentration, which varies depending on the polysaccharide chain length. 10
Typically, LPS is not considered intrinsically toxic as the endotoxic effects are primarily mediated through activation of the immune system (eg 11 ). The sensitivity to LPS depends primarily on factors altering the susceptibility of the host rather than the actual mechanisms of LPS.
LPS Sensing and Cell Activation
TLR4 Activation and the LPS Transfer Cascade
LPS that gains access to the bloodstream, for example from the gut lumen (
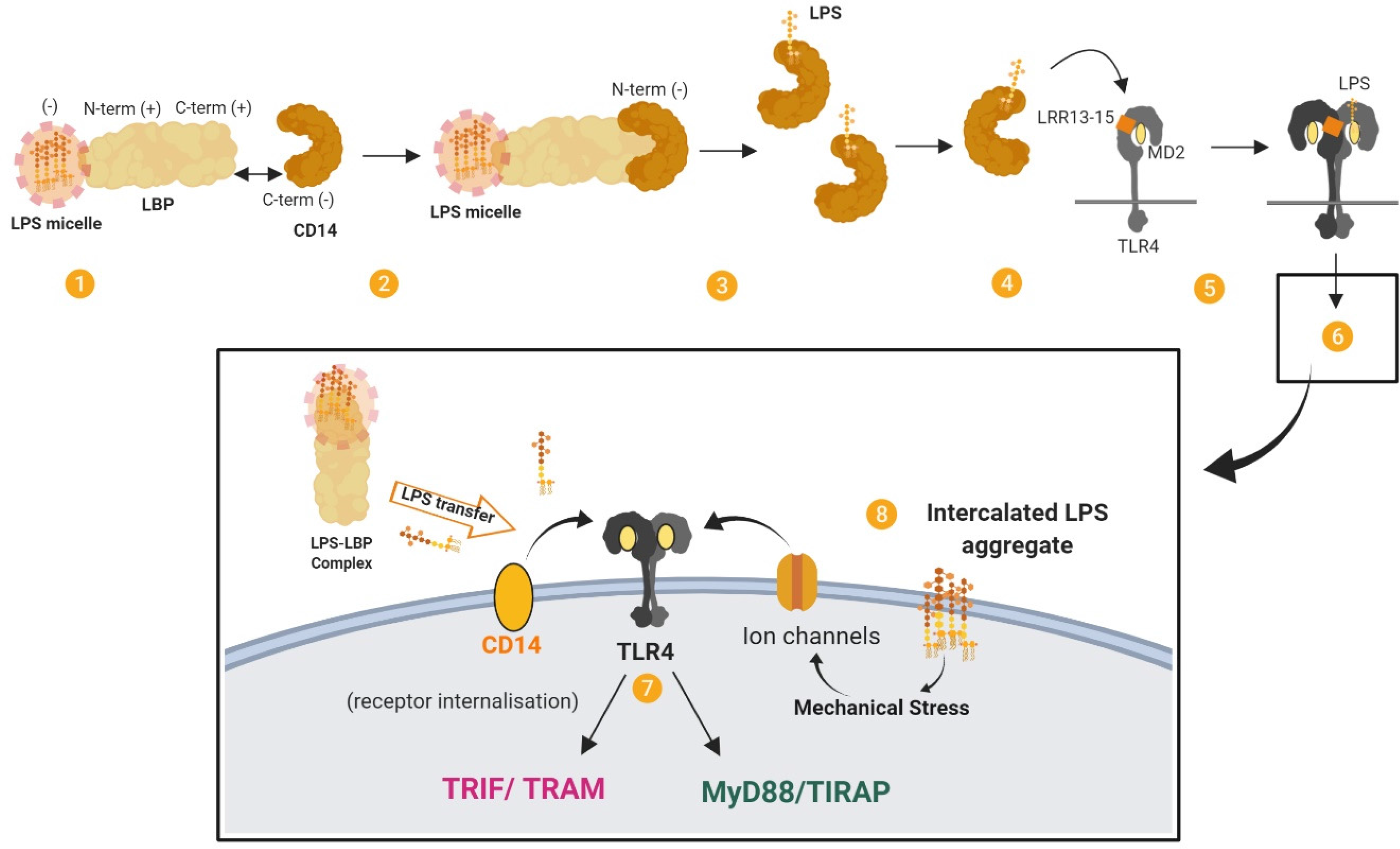
The LPS transfer cascade. The transfer of LPS to TLR4–MD2 through LBP and CD14 massively amplifies the host response to extracellular LPS. (
The TLR4–MD2 complex can be activated by structurally diverse LPS molecules. Multiple structural components of TLR4–MD2 are involved in LPS recognition, with the highly conserved lipid A portion being an important signature for recognition by the receptor complex. 16 Dimerisation of the TLR4-MD2 complex on the cell surface leads to MYD88-dependent signalling and the expression of a transcription regulators such as NFκB and activator protein 1. Subsequent internalisation of TLR4 induces MYD88-independent, TRIF-dependent signalling in endosomes and the expression of regulators such as interferon-regulatory factor 3, and eventually type 1 interferons. Ultimately this leads to the production of cytokines and chemokines such as IL-1β, IL-6, IL-8, CCL2 and TNF-α from MYD88-dependent pathways and CXCL10, CCL5, IFN-β and nitric oxide from MYD88-independent, TRIF-dependent pathways (see 17 ).
The Stimulatory Functions of LBP
LBP is an important accessory protein in the LPS transfer cascade (eg18,19 and see.15,20,21). LBP is an acute phase protein produced by hepatocytes as well as in non-hepatic tissues such as the epithelial cells of the intestine, lungs, and gingival tissue. LBP is constitutively secreted by hepatocytes into the bloodstream and circulates at low levels, but can increase from a baseline concentration of around 1 to 15 µg/mL by more than 10- to 20-fold during the acute phase response to infections.22,23
LBP can form high-affinity complexes with LPS, with a specificity for the lipid A moiety 24 (Figure 1b). LBP can extract LPS from LPS micelles (eg25,26) and bacterial membranes, 27 or LPS-rich particles like OMVs, and transfer the LPS monomers to various targets such as lipoproteins, accessory molecules and lipid domains. LBP also has an intrinsic capacity to bind to macrophage membranes in a dose-dependent manner, independently of LPS, CD14 and TLR4.10,28 Membrane-bound LBP can act as a fusion protein and enable LPS to intercalate into liposomal membrane (see 10 ). It has been proposed that membrane-bound LBP might have a role in cell activation. For example, LBP-mediated intercalation of LPS may induce mechanical stress on transmembrane proteins and stimulate K+ channels that are related to macrophage activation, with intercalated LPS aggregates being more activating than monomeric LPS.
Coreceptors and Chaperones
TLR4 can cooperate with various co-receptors, and many other molecules are involved in LPS-induced TLR4 signalling. For example, moesin is a transmembrane protein that crosslinks with the cytoskeleton and has been found to bind CD14 to form part of the complex that activates TLR4–MD2 upon LPS stimulation. 29 Furthermore, ion channels such as high-conductance Ca2+ channels and voltage-dependent K+ channels are activated by LPS in a dose-dependent manner and co-operate with TLR4 for cell activation. Here, voltage-dependent K+ channels are especially important for the NF-κB-dependent inflammatory response to LPS. 30
In terms of chaperones, extracellular high mobility group box 1 (HMGB1), like LBP, functions as an LPS transfer molecule and catalyses the transfer of LPS monomers from LPS aggregates to CD14 to initiate TLR4-mediated signalling. 31 HMGB1 binds to the polysaccharide and lipid A moieties of LPS via domains located in the A and B box domains of HMGB1, respectively. 32 HMGB1 works in concert with LPS to trigger TRIF-dependent immune responses via TLR4; downstream regulators such as receptor-interacting protein kinase 3 (RIPK3) mediate processes such as non-resolving inflammation. 33 Furthermore, HMGB1 is a ligand of receptor for advanced glycation end products (RAGE). HMGB1 acts with LPS to promote the phosphorylation of p38 MAPK and the activation of NF-κB through RAGE and thereby enhances the proinflammatory activity of LPS. 34 LPS actually stimulates the release of HMGB1 from cells such as hepatocytes in a process involving TLR4, caspase-11, and gasdermin D (GSDMD).35,36
As the function of HMGB1 suggests, cell activation can occur independently of the standard LPS transfer cascade involving LBP. For example, CD14 is dispensable for cell activation at high concentrations (100-1000 ng/mL). 37 Rough serotype LPS in particular can be recognised by the TLR4/MD-2 complex without the help of either LBP or CD14, 38 and this direct receptor activation by rough LPS leads to efficient signalling via the MYD88-dependent pathway. 39
Intracellular Sensing
In addition to its expression on the surface of various cells, TLR4 can reside on components of the endosomal systems (eg recycling endosomes, the Golgi apparatus or endoplasmic reticulum) and detect intracellular LPS independently of cell surface TLR4. 40 However, LPS can also trigger TLR4-independent responses upon intracellular detection (Figure 3).
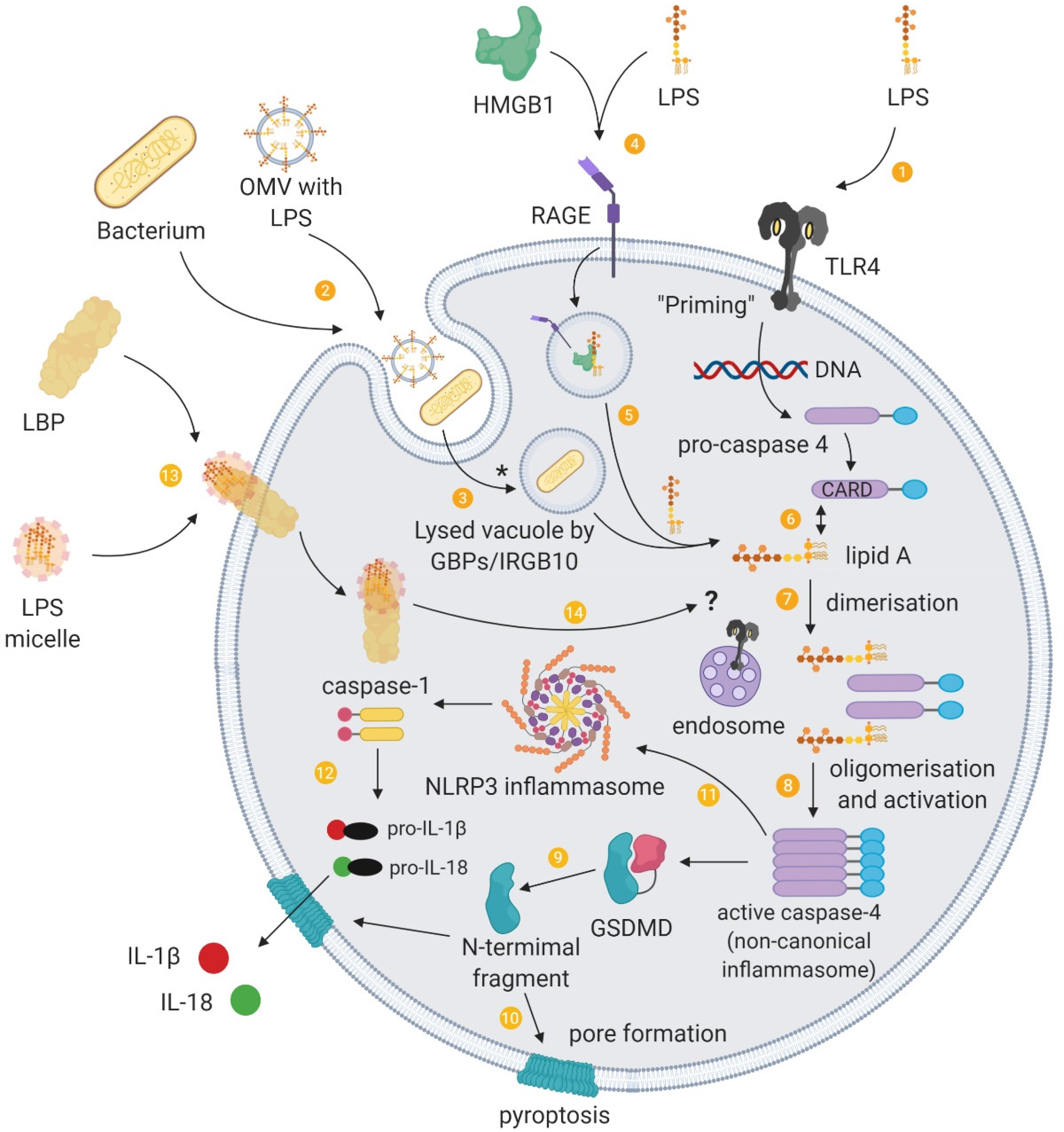
Intracellular sensing of LPS and the non-canonical inflammasome. (
LPS can access the cytoplasm by being transported into the cell via membrane-bound LBP or via HMGB1 and RAGE or by originating from the endocytosis of extracellular bacteria or their OMVs. LPS that accesses the cytoplasm is recognised and activates the non-canonical inflammasome pathway (see 41 ).
In one delivery pathway, LBP interacts with the cell membrane independently of CD14 and TLR4-MD2 and is subsequently internalised, which offers a route for intracellular LPS trafficking. 28 In addition to transferring LPS into the cytosol, LBP is found located adjacent to activated caspases. This presents a potential mechanism for the delivery of LPS to intracellular pattern recognition receptors and the noncanonical inflammasome.
As a second means of entry, extracellular HMGB1 mediates the translocation of LPS into the cytosol and facilitates caspase-11-dependent pyroptosis. 35 Specifically, HMGB1 enhances LPS internalisation into endo-lysosomes via RAGE. HMGB1 then destabilises the lysosomal membrane, allowing LPS to leak into the cytosol and activate caspase-11.
In a third pathway, pathogens and OMV can be brought into the cell through endocytosis. Pathogen-containing vacuoles (or phagosomes) carry bacterial cargo into the cell and are disrupted by interferon-inducible guanylate-binding proteins (GBPs) and IRGB10.42,43 Lysis of these vacuoles releases bacteria into the cytosol. However, GBPs do not play a role in the accessibility of OMVs to the cytosol. 44 Instead, OMVs release LPS into the cytosol from early endosomes. 45 The exact mechanism of this release is unknown, although haemolysin can induce the rupture of OMV-containing vacuoles. 46 GBPs also facilitate the sensing of cytoplasmic LPS (and OMVs44,47) by functioning as cofactors for the activation of the noncanonical inflammasome. 48 For Gram-negative bacteria that enter the cytosol, GBPs assemble on their surface and create a platform for caspase-4 activation.49,50
Caspase-4 and/or caspase-5 (caspase-11 in mice) is the primary component of the noncanonical inflammasome and acts as a cytosolic receptor for LPS.51–53 This caspase-11 pathway intersects with the extracellular TLR4 pathway. For example, the TRIF pathway, downstream of TLR4, mediates the release of type I interferons that induce caspase-11 expression 54 as well as the expression of GBPs, and is crucial for OMVs-induced caspase-11 activation. 55 Caspase-11 also synergises with the assembled canonical NLRP3 inflammasome pathway to regulate caspase-1 processing and IL-1β and IL-18 secretion. 56
An outcome of caspase-11 activation is the initiation of pyroptosis. Pyroptosis plays a role in the clearance of intracellular bacteria from macrophages, which exposes pathogens to neutrophils. GSDMD is an essential effector of pyroptosis (see57,58). GSDMD is cleaved by caspase-11 and forms pores in the cell membrane that lack ion sensitivity and compromise the integrity of cellular membranes.59–62 Cleaved GSDMD also triggers NLRP3-dependent activation of caspase-1. 59 Another upshot of this process is that NLRP3-dependent IL-1β and IL-18 could possibly exit the pyroptotic cell through the GSDMD pores.61,62
Functions of LBP in Response to LPS
LBP does not only facilitate the transfer of LPS to the TLR4 complex to maximise LPS-induced signalling (see20,21) but has multiple functions in lipid transfer and immune regulation for the recognition and control of bacterial infections. The role of LBP in these responses is concentration dependent: at low concentrations LBP potentiates LPS-induced immune activation, whereas with the acute-phase rise in LBP concentration LBP inhibits LPS-induced cellular stimulation and promotes LPS clearance to protect the host from bacterial challenge (eg63,64). As the potency of LPS is due to the sensitivity of the host recognition system, LBP has a crucial role in modulating the effects of LPS. Thus, LBP not only induces LPS responses but also terminates them (although these functions are more complex in diseases).
The Inhibitory Functions of LBP
LBP, especially at acute phase concentrations, exhibits various inhibitory actions that attenuate the stimulatory effects of LPS 23 (Figure 4). LBP can dissociate LPS from membrane-bound CD14 and inhibits the transfer of LPS from soluble CD14 and soluble MD2, thus attenuating cell signalling.64,65 LBP also promote the cellular uptake of LPS, which reduces the capacity of LPS-induced signalling through TLR4. 64 However, LPS transfer or cellular uptake facilitated by LBP is likely not the most important mechanism for clearing LPS from the blood. 66
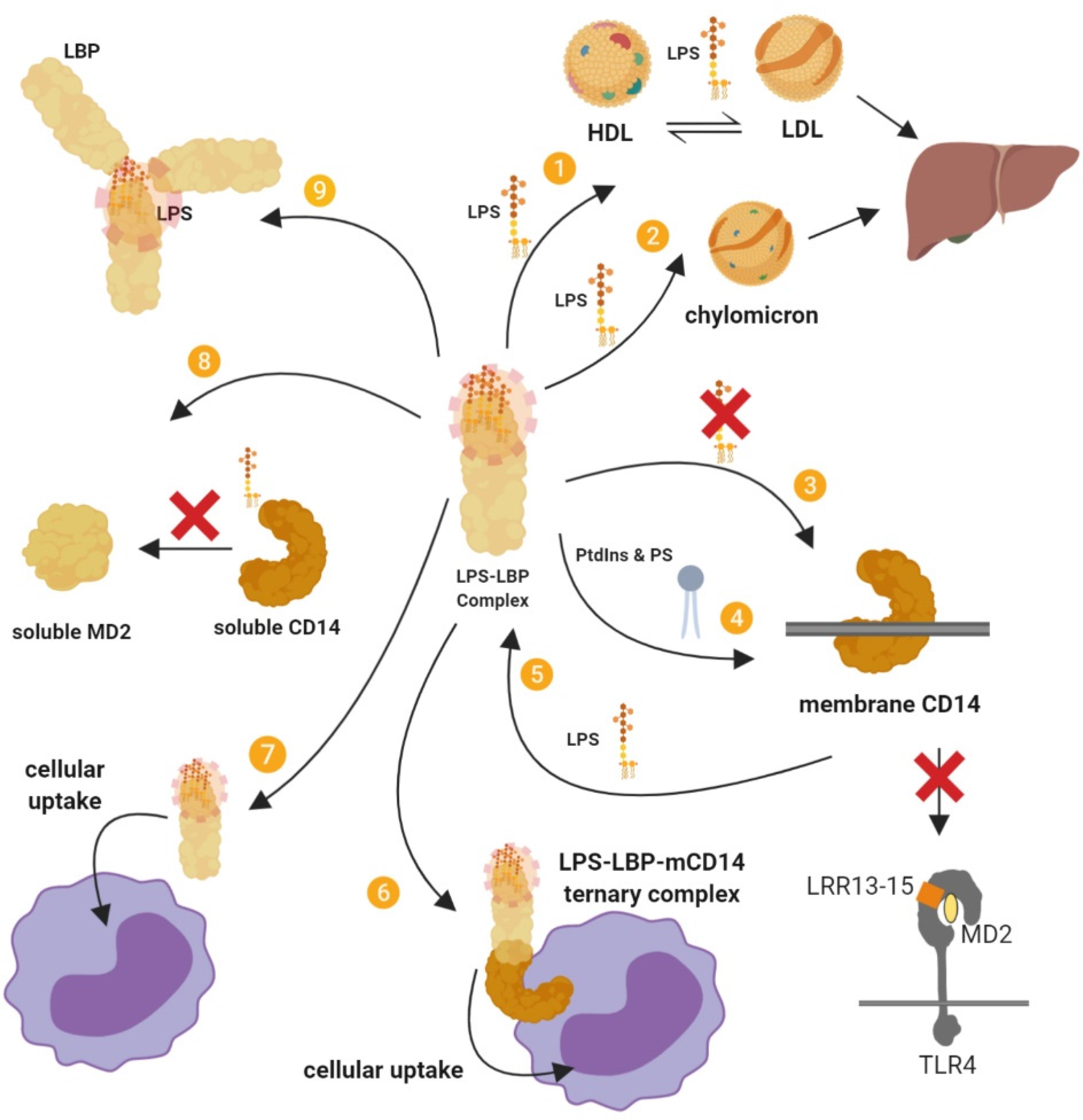
Summary of the inhibitory actions of high-dose LBP.
The inhibitory action of LBP also depends on the ability of LBP to transfer LPS to other targets including lipoproteins such as HDL27,67; a process that neutralises LPS, particularly by interfering with the internalisation of LPS by macrophages. 68 Transfer of LPS to HDL also facilitates the removal of LPS in the liver 69 , and LBP further mediate LPS detoxification in the liver by enhancing binding to chylomicrons in a dose-dependent manner. 14
Additionally, LBP intercalated into LPS aggregates can mediate the crosslinking of several layers of lipid A, which has a neutralising effect by decreasing the accessibility of lipid A. 70 This complexation increases the binding energy of the LPS in the complex as well as prevents LBP from inserting in the plasma membrane. LPS–LBP complexes in the plasma do not interact with membrane-bound LBP, which limits some of the stimulatory mechanisms of LPS. 10
Furthermore, phospholipids can inhibit the binding of LPS aggregates to LBP, probably through competitive binding, and also reduce LBP-mediated transport of LPS to the cell membrane, attenuating cell activation. 71 LBP additionally facilitates the binding of phosphatidylinositides and phosphatidylserine to membrane-bound CD14 and this inhibits LPS-induced responses in monocytes by competing for binding with LPS on CD14. 72
The Double-Edged Sword of LBP
The effects of LBP levels in disease are highlighted in Table 1. LBP is usually seen as a host-protective factor to manage the effects of LPS. 73 It enables the immune system to respond efficiently to bacterial invasion but also exhibits a dual function by blunting these effects at high doses. However, the activity of LBP in enhancing LPS-mediated stimulation and cytokine release is also viewed as a contributor to the pathogenic state mediated by LPS. In mice, antibody-mediated depletion of LBP seemingly protects against LPS-induced toxicity, 74 possibly even attenuating the onset of disease (eg diet-induced non-alcoholic fatty liver disease in mice 75 ) and dysfunction (eg endothelial dysfunction and tissue fibrosis in a swine model of LPS-induced acute kidney injury. 76 ) Furthermore, certain LBP gene polymorphisms are associated with an increased risk of pathology. For example, a functional variant in the LBP promoter (SNP 1683) has greater promoter inducibility, which is associated with higher median basal serum levels of LBP and a 5-fold increase in Gram-negative bacteraemia-related mortality of patients after allogeneic hematopoietic cell transplantation. 77 Acute-phase levels of LBP can also potentiate rather than inhibit overwhelming inflammatory responses following LPS challenge compared with LBP knockout mice in the setting of cholestatic liver disease. 78
The Role of LBP in Human Disease.

To rehearse, the reactivity of LPS depends on the sensitivity of the host and the toxic effects of LPS are mediated by the host immune system. It could be said that LBP can contribute to damaging immune reactions by augmenting immune activation. Even so, a reduced immune response in the absence of LBP results in increased susceptibility to infection and its lethal effects.21,66,79 Nonetheless LBP is not simply a protective factor.
The Role of LPS in Chronic Disease
LPS-induced signalling — through TLR4-dependent and TLR4-independent pathways — evokes a profound inflammatory response and has a wide range of effects on cells (Figure 5). In addition to the release of various inflammatory mediators following leukocyte activation (see 17 ), LPS also participates in other aspects of the immune response such as promoting dendritic cell maturation and migration, which links the innate immune response to adaptive immunity80,81 (the same is true for OMVs 82 ), and promoting autophagy in macrophages through p38 MAPK dependent TLR4 signalling, 83 which serves as a process that degrades sequestered pathogens. LPS also triggers the production of reactive oxygen and nitrogen species, such as through TLR4-mediated NADPH oxidase activation in macrophages, 84 which leads to oxidative and nitrosative stress and damage; reactive oxygen and nitrogen species are also linked to inflammatory signalling and toxic responses.

Summarised downstream effects of LPS signalling. LPS influences a range of cell types and physiological processes. The activation of leukocytes initiates the immune response and the release of various inflammatory mediators. LPS also activates specific immune processes such as the maturation and migration of dendritic cell, autophagy in macrophages, and activation of the complement system. In the liver, LPS stimulates the production of acute phase proteins as well as several inflammatory mediators. Similarly, LPS promotes inflammatory reactions in adipose tissue. These mediators as well as the increased activity of enzymes involved in the production of reactive oxygen and nitrogen species contribute to a cellular stress. A major action of LPS is its ability to promote coagulation both by enhancing the expression of molecules that stimulate clotting and through direct interaction with red blood cells and platelets. LPS-mediated changes to the endothelium also promote coagulation.
The excessive production of inflammatory mediators such as cytokines, chemokines and acute phase proteins, including complement component C3, C-reactive protein (CRP) and LBP, can result in overwhelming stimulation of the immune response and lead to cell damage and have lethal effects.3,4,85,86 This is epitomised in sepsis where a dysregulated host response to infection culminates in a pathological syndrome that can include disseminated intravascular coagulation and multisystem organ failure.4,86 However, there are many diseases in which LPS levels are raised in patients relative to matched controls.3,87 Here, the effects of LPS and its downstream signalling contribute to diverse pathologies.
Neurodegenerative Disease
LPS is implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (see 88 ). Neuroinflammation is a key pathological event in the neurodegenerative process. Systemic inflammation is similarly an important factor and systemic exposure of animal models to LPS leads to neurodegenerative pathology (see 89 ). LPS and neurodegenerative stimuli, such as β-amyloid, tau or α-synuclein, synergise to induce neurodegeneration (see88,90). LPS may prime microglia to neurodegenerative stimuli, and conversely neurodegenerative stimuli may prime microglia to LPS challenge. On the other hand, LPS exposure may lead to insensitive microglia with decreased activation, which reduces protective functions in the brain such as phagocytosis of protein aggregates. Either pathway leads to stressed neurons.
Indeed, LPS treatment of mice leads to cognitive impairment, which is prevented by TLR4-specific inhibitors.91,92 LPS also activates microglia and disrupts synapses in a mechanism involving microglia-derived IL-1β and loss of synaptophysin in mice. 93 Furthermore, LPS stimulation can increase the permeability of the blood brain barrier in aged mice. 94 Although, the blood brain barrier is relatively resistant to LPS-induced disruption, LPS can cause disruption through both paracellular and transcytotic mechanisms. 95
In the context of Alzheimer’s disease, LPS associates with amyloid plaques, neurons, and oligodendrocytes in brains of patients, and causes injury, such as to myelin.96,97 In the mouse brain, inflammation generated by systemic LPS leads to Aβ42 accumulation through increased β- and γ-secretase activities and increased expression of amyloid precursor protein, among other effects. 98 Experimentally, LPS can promote fibrillogenesis of Aβ fragments and Aβ fibrils bind directly to LPS micelles. 99 In mice, LPS-induced microglial activation also promotes hyperphosphorylation of tau through pathways involving microglial-derived IL-1 and neuronal p38 MAPK. 100
Metabolic Disease
Clinical studies of metabolic endotoxaemia in obese patients have demonstrated that serum levels of LPS increase with a high-fat diet. 101 High-fat meals similarly lead to increased LPS levels in non-obese 102 and diabetic 103 individuals. In mice, metabolic endotoxaemia dysregulates the inflammatory response and triggers the onset of diabetes and obesity. 104 Obesity is characterised as a state of chronic low-grade inflammation and LPS-induced signalling contributes to the proinflammatory milieu in human obesity. 105
LPS also promotes adipose dysfunction. LPS might be involved in defining the adipocyte death size, by initiating pyroptosis, and in the formation of crown-like structures in adipocytes, composed of dead adipocyte remnants and macrophages, as well as in promoting the transmission of macrophages to a proinflammatory phenotype (see 106 ). Indeed, obese patients with a higher degree of metabolic endotoxaemia experience altered expression in genes for adipose tissue function and lipogenesis as well as increased expression of inflammatory markers in this tissue. 107
Cardiovascular Disease
Atherosclerosis underlies the majority of cardiovascular diseases. LPS accelerates atherosclerosis in hypercholesterolemic rabbits 108 as well as provokes more severe atherosclerosis in mice. 109 Epidemiological studies show that subclinical endotoxaemia is a strong risk factor for the development of atherosclerosis. 110 Further, TLR signalling has a prominent role in atherosclerosis (see 111 ), with LPS being a potential source of vascular inflammation.
Experimentally, LPS triggers inflammatory responses in human vascular endothelial cells. 112 In studies of human tissue and primary cell culture, LPS induces the expression of neuraminidase-1, an enzyme involved in atherosclerosis progression, in monocytes, which is also part of a positive feedback loop that stimulates the expression of proinflammatory and proatherogenic cytokines. 113 Furthermore, subclinical doses of LPS in mice reduce interleukin-1 receptor associated kinase M (IRAK-M) and induce miR-24, a microRNA precursor, in monocytes, which are important for resolving inflammation and homeostatic tolerance. 114 Disruption to this system sustains the non-resolving low-grade inflammatory phenotype of monocytes, which is conducive for the aggravation of atherosclerosis.
A further way LPS experimentally contributes to atherosclerosis is by promoting lipid accumulation in human adventitial fibroblasts via TLR4 and by promoting the production of monocyte chemoattractant protein (MCP)-1, an important chemokine in the initiation of atherosclerotic plaques. 115 LPS additionally upregulates the expression of the Fcα/µ receptor on macrophages experimentally, which promotes the binding of oxidised-LDL/IgM complexes and the formation of foam cells. 116
Coagulopathy
LPS is a well-known and powerful pro-coagulant. This increases the risk for thromboembolic events, such as stoke, and can lead to more severe coagulopathy, such as disseminated intravascular coagulation. LPS upregulates the expression of tissue factor in a complement- and CD14-depedent manner, 117 which is a crucial initiator of thrombin formation and consequently fibrin deposition. LPS can also bind to fibrinogen and induce misfolding.118,119 In addition to a role in coagulation, thrombin can cleave pro-IL-1α and initiates the IL-1α inflammatory cascade. 120 Indeed, LPS-induced inflammatory processes promote coagulation (see4,86). Contrariwise coagulation factors also activate inflammatory signalling pathways, largely through protease activated receptors (see 121 ). Inflammatory processes additionally downregulate anticoagulant mechanisms such as the anti-thrombin system, tissue factor pathway inhibitor, and the activated protein C system; an increase in plasminogen activator inhibitor type 1 also contributes to curtailed fibrinolysis.
Activation of the noncanonical inflammasome by LPS is a further pathway that induces systemic blood clotting. Pyroptotic macrophages release tissue factor in the form of microvesicles through GSDMD pores, which initiates coagulation cascades. 122 Additionally, GSDMD-mediated calcium influx and activation of scramblase enzymes cause phosphatidylserine exposure that increases tissue factor activity and further drives coagulation activation. 123 Extracellular HMGB1 and its interaction with LPS for intracellular delivery promotes this process. 124 The contribution of HMGB1 to LPS-induced coagulopathy is amplified by LPS-induced type I interferon signalling downstream of TLR4 that increases the extracellular release of HMGB1. 124
Furthermore, LPS, the complement system and various inflammatory mediators induce the platelet response. 125 Platelet TLR4 activation is an important trigger for platelet haemostatic responses although the effect of LPS on platelet TLR4 is controversial (see 126 ). OMVs additionally induce platelet-platelet aggregation and degranulation as well as platelet-leukocyte binding. Ex vivo OMVs are a more potent platelet agonist than is purified LPS. 127 Platelet activation can further propagate coagulation and inflammatory signalling. 128
Therapeutic Implications
Research into therapeutic strategies that target bacterial LPS has mainly been carried out in the context of sepsis. These strategies aim to either neutralise or remove LPS or inhibit LPS-mediated activation of immune cells, thus reducing the dysregulated host response. However, therapies aimed at neutralising the effects of LPS by inhibiting the inflammatory response or by LPS-targeted antibodies (such as E5 and HA-1A) have been largely ineffective in treating sepsis. Nonetheless, it is worthwhile revising these therapeutic approaches when searching for potential therapies that target LPS in chronic diseases.
The monocolonal antibody HA-1A (marketed as Centoxin), which targets LPS, and recombinant human activated protein C (Drotrecogin alfa; marketed as Xigris), an anticoagulant, are notable failures of the biopharmaceutical industry.129,130 The LPS–TLR4 signalling pathway has also been considered as a target to treat sepsis, 131 but the lipid A antagonist eritoran, which blocks LPS from binding to TLR4–MD2, 132 and the TLR4 antagonist TAK-242 133 have been ineffective in reducing sepsis mortality. Several reasons are offered to explain the failures of these approaches including the complexity of the physiological response in stressed patients; the clinically appropriate time to administer certain drugs (eg during early exposure vs. during established pathology); heterogeneity in crucially-ill patient populations; the difficulty of standardising the timing of therapeutic interventions among patients; and the presence of generic polymorphisms within the general population that are not necessarily proportionally represented in clinical trials and potentially affect trial outcome and data analysis. 134 Of course, another explanation is simply that these reactions are not part of the main pathways driving sepsis.
Targeting LPS Through Neutralising Molecules
The development of LPS-neutralising peptides is an area of active research to treat sepsis. Protonated histidine-rich polypeptides can inhibit LPS responses by binding and neutralising LPS, although their moderate toxicity, poor solubility at physiological pH and immune activating effects limit immediate therapeutic benefit (see 135 ). Also, their LPS-neutralising effect is tightly coupled to the protonated state, which is largely lost at physiological pH. Other polycationic peptides such as procalcitonin 136 can also neutralise LPS and reduce the expression of downstream inflammatory mediators. By contrast, arginine- and lysine-rich cationic polypeptides enhance LPS responses by shuttling monomerised LPS to sites of recognition. 135
Nonetheless, peptide-based therapies based on LPS-binding domains are a promising approach to target and inhibit LPS. Peptides based on the iron-binding glycoprotein lactoferrin as well as defensins and fragments of saposin family proteins have been studied for their ability to neutralise LPS (see137,138). Synthetic anti-LPS peptides 139 and Pep19-2.5 140 also interact with LPS and interfere with its signalling cascade. Sushi peptides, derived from horseshoe crab, can bind LPS, compete against other binding proteins, and display detergent to like properties in disrupting LPS aggregates. 141 Recombinant or synthetic host antibacterial protein fragments with high affinity for LPS, such as bactericidal/permeability-increasing protein (BPI) and 18-kDa cationic antimicrobial protein (CAP18), have been proposed as candidate agents in adjunctive therapy for Gram-negative sepsis.137,138 However, problems in terms of the timing of treatment, possible toxicity, and probable rapid clearance from the circulation might limit the use of these fragments therapeutically.
Further to peptide-based therapies, other agents also demonstrate LPS neutralising abilities. Cationic lipids prevent LPS from evoking an immune response, possibly by trapping LPS in mixed micelles that are harder to disaggregate, thus detoxifying LPS. 135 Intravenous administration of protein-free, phospholipid-rich emulsions can neutralise LPS 142 but (perhaps surprisingly) these emulsions have shown no benefit in clinical trials of sepsis. 143 Lipopolyamines such as oleoyamines can also neutralise LPS and its signalling, and are of interest due to their low toxicity, low immunogenicity, and ease of chemical synthesis. 144 Lastly, synthetic cationic-amphiphilic polymers can detoxify LPS by binding and forming pseudoaggregates. 145
Other Targets to Reduce LPS and its Effects
Additional targets for treating endotoxemia might include sites of LPS release into the circulation. The gut and other focus sites (such as periodontal disease, catheters, and vascular access) are potentially reversible sources of LPS translocation into the circulation. Treatment may include the use of non-absorbable drugs that form complexes with LPS in the intestine and reduce translocation, such as sevelamer. 146 By strengthening the intestinal epithelial barrier, nutritional management including improving dietary quality and micronutrient status, manipulating the gut microbiome with prebiotics and probiotics, and using certain nutritional supplements may be alternative treatment strategies to help manage circulating levels of LPS and inflammation. 147 Indeed, natural compounds such as Zingerone from ginger attenuate LPS-induced inflammation. 148
Anti-inflammatory antioxidants may hold promise for neutralising the inflammatory effects of LPS, even if they do not interact with it directly. However, although using an anti-inflammatory therapy may seem a logical approach, research of sepsis has shown that extended immunosuppression (anergy) following an initial pro-inflammatory phase may be detrimental and increase susceptibility to secondary infections. 149 Therefore, an important consideration is to restore the balance between pro-inflammatory and anti-inflammatory reactions. This offers a further example of the complex dynamics of inflammation operating in diseases such as sepsis.
Conclusions
LPS is increasingly found to be associated with chronic diseases and can cause various pathologies. Numerous mechanisms are proposed to link LPS-induced signalling and immune dysregulation to the aetio-pathogenesis of chronic diseases. The aetiologies and pathogenesis of these diseases are multifactorial with several factors contributing to their risk and progression. LPS signalling is presented in this Review as a potential common pathway operating across these conditions.
Chronic inflammatory diseases are collectively the most prevalent conditions affecting human health, representing some of the greatest burdens to society. According to WHO factsheets, more than 55 million people have dementia (60%-70% of which is Alzheimer's disease) at an economic cost of $1.3 trillion in 2019. In 2014, 422 million people had diabetes, which accounted for 1.5 million direct deaths in 2019. Cardiovascular diseases are the leading cause of death globally at an estimated 17.9 million deaths in 2019, which represents 32% of global deaths and 38% of deaths classified as premature. Even small improvements to these statistics by regarding the role of LPS in chronic diseases could yield significant impact.
Traditionally, LPS research has been carried out in the context of sepsis, especially concerning the quest for therapeutics that target LPS. However, there exists promise for the translation of sepsis research to other pathological conditions. As an example of translation, drugs developed for sepsis treatment that improve functional outcomes (for example, by reducing morbidities, delaying the onset of organ failure, or improving outcomes on a disease-specific performance scale) but that have not been linked to reduced 28-day mortality may still show promise in other conditions where endotoxemia is less severe. Indeed, approaches that aim to reduce levels of LPS in the blood and thus mitigate its interaction with the host may lead to reduced pathology in a broad range of conditions. However, developing treatments that can lower circulating LPS levels over the long term will be challenging. Nonetheless, a starting point is to look out for and treat gut disturbances, as gut dysbiosis can be a major source for LPS to enter the blood circulation.
Overall, research in the setting of sepsis has laid a foundation for understanding LPS-induced signalling and its contribution to disease processes. Future research needs to establish the clear causal links between chronic diseases and elevated LPS. Determining the effect of therapeutics that target LPS levels or its downstream actions in chronic diseases needs to form part of these future investigations. Some success in translation has come from the failure of the antibody drug Centoxin, where experience in developing antibody therapies has informed the subsequent scientific planning and development of other antibody drugs for a number of chronic conditions (see 129 ). Appreciating the relevance of LPS-driven pathways as contributors to pathology in a range of diseases therefore offers a novel framework for approaching these conditions that might offer new solutions to the public health burden of chronic inflammatory diseases.
Footnotes
Author Contributions
M.J.P., wrote the manuscript and prepared the figures. D.B.K. reviewed and edited the manuscript. E.P. reviewed and edited the manuscript, assisted with figure design, and is the study leader.
LPS can gain entry into the body by various routes. The majority of LPS exposure arises from translocation across the gut barrier (see 166 ). The gastrointestinal tract has many features that restrain the microbiota while maintaining a symbiotic relationship (see 167 ). Disruption to the intestinal barrier and increased permeability of the gut lining enables pathogens (such as bacteria), antigens and toxins to enter the bloodstream, a state referred to as ‘leaky gut’. This can be caused by inflammatory changes that occur in various diseases (see 168 ), and is also closely associated with gut dysbiosis, an imbalance of gut microbiota (eg 169 ). Dysbiosis is implicated in the degradation and control of tight junction proteins that govern permeability of intestinal epithelial cells (eg 170 ), as well as immune dysregulation and inflammation in the intestine (see171,172). Further to this, diet, environmental stress, drug overuse, and conditions such as malnutrition or constipation may also lead to disruptions in gut barrier function and increased intestinal permeability.166,167
The bacterial product LPS has specifically been shown to translocate across the intestinal barrier and contribute to disease. LPS induces an increase in tight junction permeability through TLR4-dependent mechanisms,173–175 and contributes to immune activation and inflammation that further disrupt the gut barrier. Lipid absorption by chylomicrons also function as a vehicle for LPS entry. 13 Direct uptake of LPS may also be mediated by M cells overlaying Peyer's patches and by dendritic cells (see 3 ).
LPS may further enter the circulation at other locations, such as across compromised barriers at sites of infection. For example, LPS-induced lung inflammation is linked to increased epithelial permeability in the respiratory system. 176 Urinary tract infections can also be a source of bacterial molecules in the blood, 177 as may medical equipment such as catheters and prosthetic devices. 178 Another major source of translocated bacteria and their products is the oral cavity (see 179 ), with epithelial barriers being disrupted with abrasive toothbrushing, or periodontal disease and associated inflammation. 180 A further source of LPS is cigarette smoking (eg 181 ).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Novo Nordisk Foundation (NNF20CC0035580), UK Biotechnology and Biological Sciences Research Council (BB/L025752/1), Medical Research Council (MRC) of South Africa (Self-Initiated Research Program).
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.