
Abstract
Protease-activated receptor 2 is proposed to be a novel target for several inflammation-related diseases but its role in the central nervous system remains unclear. Protease-activated receptor 2 activation is protective in cell death and excitotoxicity assays whereas examination into the role of protease-activated receptor 2 in vivo has been hindered due to the lack of suitable pharmacological tools. Recently, a small molecule protease-activated receptor 2 activator, AC264613 (AC), was reported to be a potent and selective protease-activated receptor 2 activator that crosses the blood-brain barrier. Furthermore, peptide mimetic molecules, for example GB88, were developed that were reported to act as protease-activated receptor 2 biased antagonists. Here, we examine their signalling pathways and neuroprotective properties in central nervous system preparations. AC induced significant increases in intracellular Ca2+ in both neurons and astrocytes of primary hippocampal cultures, whereas in contrast, GB88 induced a small but significant reduction in intracellular Ca2+ in both cell types. However, both AC and GB88 induced receptor internalisation when examined using fluorescently tagged protease-activated receptor 2. Both AC and GB88 did not induce neurotoxicity in organotypic hippocampal slice cultures when applied alone but reduced neurotoxicity when co-applied with kainate in excitotoxicity assays. Furthermore, both AC and GB88 reduced neurotoxicity when applied post kainate insult indicating they exhibit neuroprotective properties even after excitotoxicity is induced. These data indicate that protease-activated receptor 2 activation is neuroprotective but this is independent of Gq-induced Ca2+ activation. Given that AC crosses the blood–brain barrier, this highlights its use as a novel tool to examine the protective properties of protease-activated receptor 2 in in vivo models of central nervous system disorders.
Keywords
Introduction
Protease-activated receptors (PARs) are G-protein coupled receptors (GPCRs) that have a unique activation mechanism by which proteases cleave the N-terminus and reveal a ‘tethered ligand’ that binds to the second extracellular loop leading to activation of signalling pathways (McIntosh et al., 2020; Ramachandran et al., 2012). One PAR subtype, PAR2, has been linked to inflammatory responses and is proposed to be a novel target for several inflammation-related diseases including cancer, irritable bowel disease and rheumatoid arthritis (Khoon and Piran, 2025; Shah et al., 2025; Villano and Pontisso, 2024). PAR2 is also proposed to play a role in neurogenic inflammation and associated pain pathways and recently PAR2 has been targeted as a treatment for migraine (Kopruszinski et al., 2025; Mason et al., 2023). However, the role of PAR2 in the central nervous system (CNS) remains unclear.
We and others have shown that PAR2 activation, using peptides that mimic its tethered ligand, induces increases in intracellular Ca2+ in neurons and astrocytes of primary hippocampal cultures (Bushell et al., 2006; Wang et al., 2002) and modulates synaptic transmission in a region-specific manner (Gan et al., 2011; Mrozkova et al., 2021; Shavit-Stein et al., 2017). Furthermore, PAR2 activation is protective against ceramide-induced cell death (Wang et al., 2007a, 2007b) and is neuroprotective against excitotoxicity induced in organotypic slice cultures (OSCs; Greenwood and Bushell, 2010). However, examination into the role of PAR2 in vivo was hindered due to the lack of suitable pharmacological tools (McIntosh et al., 2020; Ramachandran et al., 2012). Despite this, it was shown that lipopolysaccharide (LPS)-induced sickness-like behaviour was impaired in PAR2 deletion mice but did not alter behaviour per se (Abulkassim et al., 2016). Furthermore, changes in PAR2 expression have been shown in several inflammation-related CNS disorders (Afkhami-Goli et al., 2007; Hurley et al., 2015; Noorbakhsh et al., 2005, 2006) but whether this plays either a detrimental or protective role is disease-dependent (Bushell, 2007) and requires suitable PAR2 activators and inhibitors to examine this further. To this end, a small molecule PAR2 activator, AC264613 (AC), was reported to be a potent and selective PAR2 activator (Gardell et al., 2008), which crosses the blood–brain barrier (BBB) and induces depression-like behaviour in mice (Moudio et al., 2022). Peptide mimetic molecules, for example, GB88, were also developed that were reported to act as PAR2 biased antagonists (Suen et al., 2012) and impair the inflammatory actions of PAR2 activators (Francis et al., 2023; Lieu et al., 2016).
With the advent of these new pharmacological tools, we examined their action on Ca2+ signalling and receptor internalisation as well as determining their neuroprotective properties in an in vitro excitotoxicity assay in comparison to known peptide PAR2 activators. We found that effects on Ca2+ signalling were drug-dependent but all compounds induced receptor internalisation. PAR2 activators were not neurotoxic to organotypic hippocampal slice cultures (OHSCs) but all reduced neurotoxicity when co-applied with kainate (KA) in excitotoxicity assays. These data indicate that PAR2 activation is neuroprotective but suggest that this is independent of Gq-induced Ca2+ activation.
Materials and methods
Cell culture
tsA201 cell culture and transfection: tsA201 cells (ECACC catalogue no. 85120602) were maintained in growth media containing Dulbecco’s Modified Eagle Medium (DMEM, Cat No: 21969035), 10% foetal calf serum (Cat No: A5256701), 1% non-essential amino acids (Cat No: 11140050) and 1% penicillin (10,000 U/mL) streptomycin (10 mg/mL; Cat No: 15140122) in a humidified incubator at 37°C/5% CO2. When the cells were at least 90% confluent, they were split and plated for transfection onto poly-
Primary hippocampal cultures: Primary hippocampal cultures were prepared as described previously (Hridi et al., 2019). Briefly, Sprague-Dawley pups (1–2 days old) were sacrificed by cervical dislocation and decapitated in accordance with UK Home Office Schedule 1 guidelines under the authority of UK Animals (Scientific Procedures) Act, 1986. Under sterile conditions, the brain was removed and the hippocampi dissected out and placed in dissection solution containing (in mM) NaCl 116, KCl 5.4, NaHCO3 26, NaH2PO4 1.3, MgSO4 2, glucose 25, CaCl2 2 (all Sigma, UK), with pH adjusted to 7.4. The hippocampi were then incubated for 20 min in papain solution (1.5 mg/mL; Cat No: P4672 in dissection solution), following which they were transferred to dissection solution containing BSA (1%, Cat No: A3294), triturated with flame polished pasteur pipettes and the resulting dissociated cells were plated at a density of 3 ×105 cells/mL onto poly-
OHSCs: OHSCs were prepared similar to previously described (Greenwood and Bushell, 2010; McCarthy et al., 2021). Briefly, C57BL/6 J mice pups (5–8 days old) were obtained from Harlan UK Ltd and killed by cervical dislocation and decapitation according to UK Home Office Schedule 1 guidelines under the authority of UK Animals (Scientific Procedures) Act, 1986 and approved by the Eli Lilly Animal Welfare Board. In a laminar flow hood, the head was sprayed with ice-cold 70% ethanol and the brain rapidly removed and placed in filter sterilised ice-cold dissection buffer (Gey’s balanced salt solution (98.5%; Cat No: G9779),
Drugs
All compounds were made as 1000x stock solutions and diluted accordingly as required. KA and kynurenic acid (KYN) were made in deionised water and stored in a −20°C freezer until required. AC264613 (AC) stock solutions were made in dimethyl sulfoxide (DMSO), stored in a −20°C freezer with the final bath concentration of DMSO being a maximum of 0.1% in all experiments. All PAR2 peptide activators were made fresh each day in phosphate-buffered saline (PBS). DMSO (0.1%) and PBS (0.1%) were used as vehicle controls where appropriate, with these combined as vehicle controls as both were without effect in any experiments. Drug concentrations were chosen based our previous studies (Gan et al., 2011; Greenwood and Bushell, 2010; Shrestha et al., 2014).
Quantification of receptor internalisation
TsA201 cells were washed with warmed serum-free DMEM 24–48 h post-transfection, following which 2-mL serum-free DMEM containing the relevant concentration of the drug of interest (AC, Tocris Bioscience Cat No: 3370-50), GB88 kindly produced by Dr C Jamieson, University of Strathclyde, SLIGRL-NH2 obtained from Peptide Synthetics, UK) was added and the cells placed in the incubator for 45 min. Coverslips were then transferred to a HEPES-based buffer containing in mM: NaCl 140, KCl 2.5, MgCl2 2, HEPES 10,
Ca2+ imaging
Ca imaging was performed as previously described (Hridi et al., 2019). Hence, 11–14 DIV, primary hippocampal cultures were washed with HEPES-buffered saline (HBS) composed of the following (in mM): NaCl 140, KCl 5, MgCl2 2, CaCl2 2, HEPES 10,
OHSC neurotoxicity assay
At 14 DIV, OHSCs were treated with propidium iodide (PI, 3 µM, 3 h) and imaged to examine health and viability using a BD pathway 855 High-Content Bioimager (BD Biosciences, UK) as described previously (McCarthy et al., 2021). For neurotoxicity assays, experimental compounds were added directly to fresh culture media at the concentrations indicated and equilibrated at 37°C/5% CO2 before use. Slices exposed to compounds were maintained at 37°C/5% CO2 for the duration of experimental treatment with PI fluorescence analysed using the BD Bioimager. For 24 h treatments, PI was included in the media throughout, whereas for 1 h treatments, fresh media containing PI replaced the experimental treatment and slices were imaged 24 h later. For consistency, identical image settings were used in all experiments with all toxicity data expressed as a percentage of the maximum PI fluorescence observed following exposure to kainic acid (300 µM, 24 h) with n = OHSC data obtained from ⩾2 slices per animal from which they were prepared.
Statistical analysis
All data are expressed as mean ± S.E.M. with differences examined for statistical significance using one-way ANOVAs with Dunnett’s or Tukey’s post hoc test where appropriate using Minitab (v19), with p < 0.05 taken as significant.
Results
AC and GB88 induce opposite effects on intracellular calcium levels in primary hippocampal cultures
AC (Gardell et al., 2008) and GB88 (Suen et al., 2012) have been reported to be a selective PAR2 agonist and antagonist, respectively. Here, we examined whether they modulate neuronal intracellular calcium (n[Ca2+]i) levels or astrocytic intracellular calcium (a[Ca2+]i) levels in primary hippocampal cultures as we have shown previously for the peptide activator SLIGRL (Bushell et al., 2006; Gan et al., 2011). Application of AC (50 µM) significantly increased n[Ca2+]i by 0.77 ± 0.0.09 (n = 6, p < 0.001 vs vehicle control, Figure 1(c) and (d)) and a[Ca2+]i by 0.92 ± 0.07 (n = 6, p < 0.001 vs vehicle control, Figure 1(d)). Similarly, SLIGRL-NH2 (50 µM) increased n[Ca2+]i by 0.84 ± 0.06 (n = 7, p < 0.001 vs vehicle control, Figure 1(c) and (d)) and a[Ca2+]i by 0.97 ± 0.07 (n = 7, p < 0.001 vs vehicle control, Figure 1(d)). In contrast, GB88 (50 µM) reduced n[Ca2+]i by 0.10 ± 0.02 (n = 7, p < 0.05 vs vehicle control, p < 0.001 vs SLIGRL, Figure 1(d)) and a[Ca2+]i by 0.17 ± 0.02 (n = 7, p < 0.01 vs vehicle control, p < 0.001 vs SLIGRL, Figure 1(d)).

PAR2 activators induce receptor internalisation but have differing effects on intracellular Ca2+. (a) Representative Ca2+ imaging traces of SLIGRL and AC induced increases in intracellular Ca2+. (b) Bar chart summarising the effects of PAR2 modulators on [Ca2+]i in astrocytes and neurons of rat primary hippocampal cultures. Data are changes in fluorescence ratio from the normalised baseline with n = mean data from ⩾ 5 cells from each culture obtained from different animals. * = p < 0.05, *** = p < 0.001 versus vehicle control, ### = p < 0.001 versus SLIGRL, one-way ANOVA with Tukey’s post hoc test. (c) Representative images of PAR2-GFP expression in tsA201 cells in the absence (vehicle = DMSO) and presence of PAR2 activators. (d) Bar chart revealing that all PAR2 modulators induce internalisation compared to vehicle controls. All data was acquired ⩾ 5 cells from at least 5 separate transfected tsA201 cultures. ***= p < 0.001 versus vehicle control, one-way ANOVA with Dunnett’s post hoc test. Scale bar = 10 µm.
AC-264613 and GB88 both induce PAR2 internalisation
Having established that AC increases but GB88 reduces [Ca2+]i in primary hippocampal cultures, we examined their effect on PAR2 activation by examining receptor internalisation in tsa201 cells. GB88 (50 µM) increased the PAR2-GFP internalisation ratio to 0.43 ± 0.02 (n = 5, p < 0.001 vs vehicle control, Figure 1(a)) from a vehicle control internalisation ratio of 0.28 ± 0.01 (n = 7, Figure 1(a) and (b)). Strikingly, these values were similar to that induced by the PAR2 activating peptide, SLIGRL-NH2 (50 µM, 0.47 ± 0.01, n = 9, p < 0.001 vs vehicle control, Figure 1(b)) and AC (50 µM, 0.47 ± 0.01, n = 9, p < 0.001 vs vehicle control, Figure 1(b)).
AC and GB88 are not neurotoxic to OHSCs
Having confirmed that both AC and GB88 activate PAR-2 resulting in receptor internalisation but have differing effects on [Ca2+]I levels in primary hippocampal cultures, we next examined whether exposure to these PAR2 activators induced neurotoxicity in OHSCs. In agreement with our previous findings (Greenwood and Bushell, 2010), exposure to SLIGRL-NH2 (50 µM, 24 h) did not induce neurotoxicity in OHSCs (22.4 ± 1.3%, n = 6, Figure 2(a) and (b)) when compared to vehicle controls (20.9 ± 2.3%, n =, Figure 2(a) and (b)), whereas a positive control, kainic acid (KA, 300 µM, 24 h), resulted in 100 ± 1.5% cell death (n = 6, p < 0.001 vs vehicle control, Figure 2(a) and (b)). Similar to SLIGRL-NH2, both AC (21.7 ± 0.8%, n = 6, Figure 2(a) and (b)) and GB88 (21.2 ± 1.1%, n = 6, Figure 2(a) and (b)) did not induce toxicity when compared to vehicle controls.

PAR2 activators do not induce neurotoxicity in OHSCs. (a) Representative images of propidium iodide fluorescence in OHSCs 24 h after exposure to KA, SLIGRL, AC and GB88. (b) Bar chart illustrating that PAR2 activators are not neurotoxic in OHSCs when compared to KA-induced neurotoxicity. Data are expressed as a percentage of the maximum PI fluorescence observed following exposure to kainic acid (300 µM, 24 h) with n = OHSC data obtained from ⩾2 slices per animal from which they were prepared. *** = p < 0.001 versus vehicle control, one-way ANOVA with Dunnett’s post hoc test. Scale bar = 250 µm.
AC and GB88 are neuroprotective against KA-induced neurotoxicity
Having established that AC and GB88 are not neurotoxic to OHSCs, we next investigated whether these PAR2 activators were neuroprotective against KA-induced neurotoxicity. KA (300 µM, 24 h) resulted in 100 ± 3.6% cell death (n = 7, p < 0.001 vs vehicle control, Figure 3(a) and (b)), which was significantly inhibited by the broad spectrum glutamate antagonist, kynurenic acid (KYN (3M), 32.3 ± 3.1%, n = 6, p < 0.001 vs KA (300µM, 24 h), Figure 3(b)). When co-applied with KA (300 µM, 24 h), SLIGRL-NH2 (48.6 ± 2.0%, n = 5, p < 0.001 vs KA (300 µM) alone, Figure 3(a) and (b)), AC (42.7 ± 1.7%, n = 6, p < 0.001 vs KA (300 µM) alone, Figure 3(a) and (b)) and GB88 (44.3 ± 1.4%, n = 5, p < 0.001 vs KA (300 µM) alone, Figure 3(a) and (b)) all significantly inhibited KA-induced neurotoxicity. In addition, we examined whether PAR2 activation was protective against neurotoxicity induced by lower KA concentrations. Indeed, SLIGRL-NH2 (23.2 ± 2.0%, n = 6, p < 0.001 vs KA (20 µM) alone, Figure 3(c)), AC (22.3 ± 2.0%, n = 5, p < 0.001 vs KA (20 µM) alone, Figure 3(c)) and GB88 (22.9 ± 0.7%, n = 5, p < 0.001 vs KA (20 µM) alone, Figure 3(c)) significantly inhibited neurotoxicity when co-applied with KA compared to KA alone (20 µM, 24 h, 52.5 ± 1.2% of max cell death, n = 5). As expected, in the presence of KYN (3M), KA-induced neurotoxicity was reduced 13.5 ± 3.0%, (n = 5, p < 0.001 vs KA (20 µM) alone, Figure 3(c)).

PAR2 activators are neuroprotective when co-applied with KA in OHSCs. (a) Representative OSHC images illustrating that PAR2 activators reduce neurotoxicity when co-applied with KA (300 µM, 24 h). (b) Summary bar chart highlighting that SLIGRL, AC and GB88 reduce neurotoxicity when co-applied with KA (300 µM, 24 h). ***= p < 0.001 versus KA alone, one-way ANOVA with Dunnett’s post hoc test. (c) OHSC neurotoxicity is reduced when SLIGRL, AC and GB88 are co-applied with KA (20 µM, 24 h). Data are expressed as a percentage of the maximum PI fluorescence observed following exposure to kainic acid (300 µM, 24 h) with n = OHSC data obtained from ⩾ 2 slices per animal from which they were prepared. ***= p < 0.001 versus KA alone, one-way ANOVA with Dunnett’s post hoc test. Scale bar = 250 µm.
PAR2-mediated neuroprotection remains post-neurotoxic insult
To more reliably reflect what would be required of a neuroprotective treatment when examined in in vivo animal models as well as patients with neurodegenerative diseases, we next examined whether PAR2 activation was neuroprotective if applied after KA-induced neurotoxicity was initiated. KA (300 µM, 1 h) resulted in 71.8 ± 3.6% of max cell death (n = 5, Figure 4(b)), with KYN (3M) having no effect (68.6 ± 2.4% of max cell death, n = 5, Figure 4(b)) when applied to OHSCS for 23 h following removal of KA (300 µM). In contrast to the lack of KYN neuroprotection, SLIGRL-NH2 (56.4 ± 1.0%, n = 5, p < 0.001 vs KA (300 µM) alone, Figure 4(b)), AC (52.6 ± 1.1%, n = 6, p < 0.001 vs KA (300 µM) alone, Figure 4(b)) and GB88 (56.2 ± 1.6%, n = 5, p < 0.001 vs KA (300 µM) alone, Figure 4(b)) significantly reduced neurotoxicity when applied for 23 h post OHSC exposure to KA (300 µM, 1 h). Similarly, AC (14.5 ± 0.5%, n = 5, p < 0.01 vs KA (20 µM) alone, Figure 4(c)) and GB88 (15.7 ± 0.6%, n = 4, p < 0.05 vs KA (20 µM) alone, Figure 4(c)) significantly impaired neurotoxicity when applied following OHSC exposure to KA (20 µM, 1 h, 20.1 ± 1.5%, n = 5) whereas post-application of KYN (3M, 20.4 ± 1.4%, n = 5) and SLIGRL-NH2 (16.1 ± 1.1%, n = 5, Figure 4(c)) was ineffective.
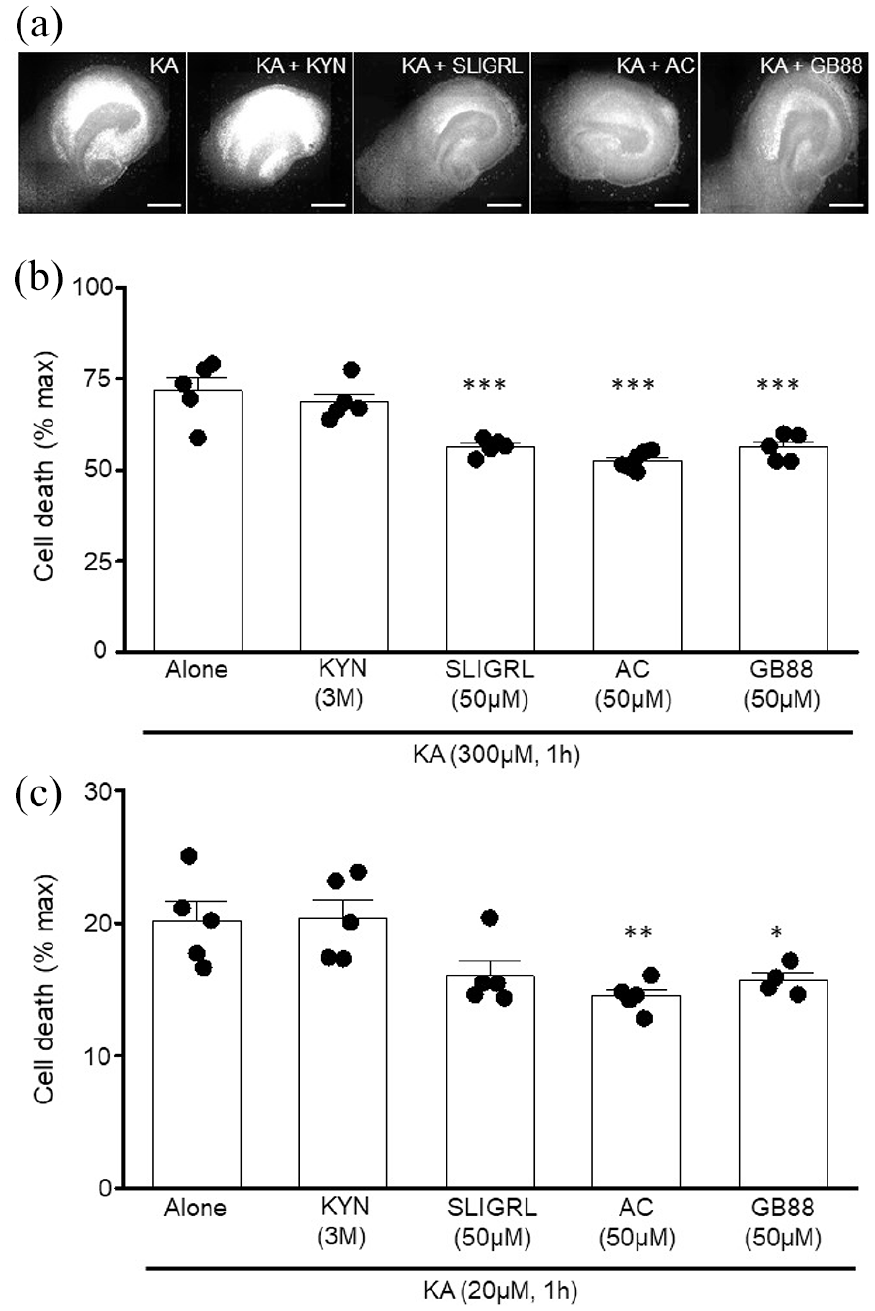
PAR2 activators are neuroprotective even after initiation of KA-induced neurotoxicity. (a) Representative images illustrating that PAR2 activators reduce neurotoxicity when applied after exposure to KA (300 µM, 1 h). (b) Bar chart revealing that KA-induced neurotoxicity is reduced even when SLIGRL, AC and GB88 are applied after OHSC exposure to KA (300 µM, 1 h). ***= p < 0.001 versus KA alone, one-way ANOVA with Dunnett’s post hoc test. (c) Summary showing that AC and GB88 reduced OHSC neurotoxicity when applied following KA (20 µM, 1 h) application. Data are expressed as a percentage of the maximum PI fluorescence observed following exposure to kainic acid (300 µM, 24 h) with n = OHSC data obtained from ⩾ 2 slices per animal from which they were prepared. *= p < 0.05, **= p < 0.01 versus KA alone, one-way ANOVA with Dunnett’s post hoc test. Scale bar = 250 µm.
Discussion
PAR2 is proposed to be a novel target for certain CNS disorders but whether new PAR2 pharmacological tools affect CNS cell signalling and are neuroprotective is unknown. Here, we show that PAR2-induced Ca2+ signalling is drug-dependent whereas all drugs tested induce receptor internalisation. In addition, they are all neuroprotective in excitotoxicity assays.
We and others have previously shown that PAR2 activation elicits increases in [Ca2+]i in both primary cultured astrocytes and neurons (Bushell et al., 2006; Gan et al., 2011; Wang et al., 2002). In the present study, we show for the first time that the small molecule PAR2 activator, AC264613, increases astrocytic and neuronal [Ca2+]i in primary hippocampal cultures similar to that observed with established peptide PAR2 activator, SLIGRL-NH2. Our findings are in agreement with previous studies that revealed that AC induces increased [Ca2+]i in KNRK cells transiently transfected with PAR2 (Gardell et al., 2008) and human bronchial and epithelial cells (Ocasio-Rivera et al., 2020) and that PAR2 activation using peptide activators increases [Ca2+]i in numerous cell types (Macfarlane et al., 2001; Zhuo et al., 2022). In contrast, the proposed PAR2 biased antagonist, GB88 (Suen et al., 2012), induced a small but significant reduction in [Ca2+]i in both primary hippocampal astrocytes and neurons. Previous studies have indicated that pre-exposure to GB88 impairs PAR2 activator induced increases in [Ca2+]i (Suen et al., 2012, 2014; Wang et al., 2021) but can also induce cAMP, ERK and Rho signalling in human cell lines, thus being deemed to have pathway-selective antagonism (Suen et al., 2014). Furthermore, given GB88’s oral availability, it has been shown to reduce inflammation related pathways in several in vivo studies (Francis et al., 2023; Liao et al., 2025; Lieu et al., 2016). However, whether GB88 activation of these pathways underlies the observed reduction of astrocytic and neuronal [Ca2+]i is beyond the scope of these studies. However, both AC and GB88 do induce PAR2 internalisation in tsa201 cells similar to that induced by SLIGRL-NH2, confirming previous studies that both are PAR2 activators but do so in a pathway-dependent manner (Gardell et al., 2008; Suen et al., 2014). Whether receptor internalisation would be induced by these compounds in primary hippocampal cultures, as has been done for other GPCRs (Bushell et al., 2002), would be important to confirm but is beyond the scope of this study.
Having examined Ca2+ signalling following exposure to AC and GB88 in primary hippocampal cultures, we then examined whether PAR2 activation is neuroprotective in an OHSC excitotoxicity assay. Initial experiments revealed that activating PAR2 with AC, GB88 and SLIGRL-NH2 for 24 h does not induce neurotoxicity in OHSCs when examined using PI fluorescence. This is in agreement with our previous findings in OHSCs (Greenwood and Bushell, 2010) but in contrast to findings when examined in primary hippocampal cultures (Smith-Swintosky et al., 1997). Whether these differences are due to the preparations used is unknown. As OHSCs are a more physiologically relevant preparation, the lack of toxicity in them suggests this would be the case in vivo, especially as the pharmacokinetics for the only BBB-permeable activator, AC, reveal that it is present in the brain for only a short period (Moudio et al., 2022). Given the absence of neurotoxicity following extended PAR2 activation, we examined whether co-application of PAR2 activators impaired KA (300 µM, 24 h)-induced neurotoxicity. Indeed, co-application of all three PAR2 activators significantly reduced KA-induced neurotoxicity as did the ionotropic glutamate receptor antagonist, kynurenic acid. Furthermore, PAR2 activation with AC, GB88 and SLIGRL-NH2 significantly reduced neurotoxicity induced by KA (20 µM, 24 h), a concentration chosen to mimic the slower developing excitotoxicity observed in certain CNS conditions including stroke. These data are in agreement with our previous study in OHSCs in which we revealed that PAR2 activation with SLIGRL-NH2 was neuroprotective (Greenwood and Bushell, 2010) and other studies that have indicated that PAR2 can play a protective role against experimentally-induced apoptosis (Wang et al., 2007a), experimental ischaemia (Jin et al., 2005) and HIV-associated dementia (Noorbakhsh et al., 2005). We also extended our investigation to examine whether of PAR2-induced neuroprotection was evident after the initiation of KA-induced neurotoxicity. Exposing OHSCs to AC and GB88 post KA-induced neurotoxicity remained neuroprotective under both conditions revealing that PAR2 activation is neuroprotective even if initiated post insult. While our study utilises an assay to mimic the neurotoxicity observed in the CNS, the fact that AC and GB88 continued to provide protection post KA insult more accurately reflects how a neuroprotective treatment would be administered in the event of a CNS insult in vivo. Having shown that AC can cross the BBB and that it has a short half-life in the brain (Moudio et al., 2022), then AC is a good candidate to examine whether the in vitro protection observed here translates to animal models of CNS disorders. In hindsight and given the focus on credibility in neuroscience, we acknowledge that if the control and the neuroprotective experiments were carried out simultaneously using OHSCs prepared at the same time, then two-way ANOVAs could have been used and this would have improved the robustness of our findings. In addition, use of other assays, for example, LDH assays to detect cytotoxicity, NeuN staining for slice architecture, would have confirmed the health of our OHSCs and the neuroprotective effects of PAR2 activation.
We have shown previously that PAR2 modulation of synaptic transmission (Gan et al., 2011) and neuroprotection in excitotoxicity assays (Greenwood and Bushell, 2010) is mediated via astrocytes. While not a focus of the current study, given that AC signalling mechanisms are identical to those of SLIGRL-NH2, it is likely that AC-induced neuroprotection is also astrocytically mediated but whether this is the case for GB88 requires further investigation. Furthermore, given that both AC and GB88 are neuroprotective despite activating PAR2 in a pathway-dependent manner, the exact signalling mechanisms underlying the observed neuroprotection remains unclear. AC induced increases in [Ca2+]i in primary hippocampal cultures is in agreement with numerous previous studies highlighting that PAR2 is a GPCR that primarily signals via the Gq-PLC pathway (McIntosh et al., 2020; Reches and Piran, 2024). With Gq GPCR activation been shown to be protective in numerous studies (Tobin, 2024), this may underlie the AC-induced neuroprotection. In contrast, GB88 does not signal via the Gq-PLC pathway in primary hippocampal cultures but has previously been shown to increase ERK signalling in several cell types (Park et al., 2009; Suen et al., 2014; Wang et al., 2021). However, whether increased ERK signalling, which has previously been reported for G-protein independent PAR2 signalling (McIntosh et al., 2020; Ramachandran et al., 2012) underlies the GB88-induced neuroprotection remains to investigated but it should be noted that we have shown previously that in OHSCs, PAR2 activation results in a decrease in both p38 MAPK and ERK signalling (Greenwood and Bushell, 2010). However, despite the exact mechanisms remaining unclear, this study, alongside previous studies examining the role of PAR2 in CNS disorders, highlights that PAR2 is a potential therapeutic target for the treatment of CNS disorders.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a University of Strathclyde PhD CASE studentship with Eli Lilly Ltd to SM.