
Abstract
Maternal obesity is associated with the development of a variety of neuropsychiatric disorders; however, the mechanisms behind this association are not fully understood. Comparison between maternal immune activation and maternal obesity reveals similarities in associated impairments and maternal cytokine profile. Here, we present a summary of recent evidence describing how inflammatory processes contribute towards the development of neuropsychiatric disorders in the offspring of obese mothers. This includes discussion on how maternal cytokine levels, fatty acids and placental inflammation may interact with foetal neurodevelopment through changes to microglial behaviour and epigenetic modification. We also propose an exosome-mediated mechanism for the disruption of brain development under maternal obesity and discuss potential intervention strategies.
Keywords
Introduction
During the extremely dynamic and plastic foetal period, neural cells proliferate, migrate and generate the blueprint for functional neuronal circuits. Importantly, recent advances in neurosciences and psychiatry point to an early origin of neuropsychiatric disorders. This early life period appears to be highly sensitive to both genetic and environmental stress. Early life stressors are thought to impair these processes to ‘prime’ the brain for later psychiatric disorders, extending the Developmental Origin of Health and Disease (DOHaD) concept to brain disorders. Accordingly, large-scale population studies demonstrate that maternal obesity increases the risk for neuropsychiatric disorders in offspring (Rivera et al., 2015). These increased risks apply to neurodevelopmental disorders (ASD: autism spectrum disorders, DD: developmental delay, ID: intellectual disability and ADHD: attention deficit hyperactivity disorder) (Heikura et al., 2008; Krakowiak et al., 2012; Rodriguez et al., 2008) but also to schizophrenia and depression (Edlow, 2017). These epidemiological findings are supported by studies in animal models of maternal obesity, in which behavioural deficits in offspring recapitulate the main features of these disorders. Given the current obesity pandemic – 650 million adults obese in 2016 and 340 million children between 5 and 19 overweight or obese (World Health Organization (WHO)) – and continuously rising trends, understanding the mechanisms underlying the transgenerational effect of maternal obesity on the developing brain is a major challenge.
Neuroinflammation, including in early life, recently gained much interest in neuropsychiatry, providing new pathophysiological mechanisms and hope for new treatments (Brown and Derkits, 2010; Brown and Meyer, 2018; Gumusoglu and Stevens, 2019). Because obesity is associated with chronic low-grade inflammation, exploring a neuroinflammatory hypothesis for the increased risks of mental diseases in the offspring of maternal obesity is appealing. In this review, we will present evidence for inflammation during pregnancy while obese and compare findings in animal models of maternal obesity and maternal immune activation (MIA). We will then discuss how this might deregulate the foetal immune system and ‘prime’ the developing brain with a particular focus on microglia, the resident brain macrophages. The role of microbiota, a possible link between inflammation and some neurobehavioral outcomes, will not be discussed here as this highly debated topic would deserve a full article. Interested readers are referred to recent comprehensive reviews (Codagnone et al., 2019; Vuong and Hsiao, 2017).
Behavioural outcomes in animal models of maternal obesity and maternal infection
Infection during pregnancy is associated with increased risk for neurodevelopmental and neuropsychiatric disorders including ASD, schizophrenia, childhood depression and bipolar disorders (Brown and Meyer, 2018; Estes and McAllister, 2016). While associations between maternal obesity and mental disorders overlap with those of infection (e.g. ASD or schizophrenia) (Edlow, 2017), respective risks likely differ, although no studies directly compared the two maternal conditions. These epidemiological findings are supported by a number of preclinical studies using animal models of maternal high-fat diet (MHFD) and MIA. We will focus here on recent findings in rodent models, although behavioural alterations have been identified in several other animal models including non-human primates (Thompson et al., 2017, 2018). A common challenge to MHFD and MIA models resides in differences in methodologies and designs including the use of bacterial (LPS, lipopolysaccharide) or viral (Poly: IC, Polyinosinic: polycytidylic acid) mimics for MIA, high-fat or high-fat/high-sugar diets, concentrations and timing, as well as behavioural testing setups. In addition, extrapolating performance of rodents in behavioural testing to psychiatric phenotypes, in particular mood and social behaviours, is limited as it does not faithfully replicate the complexity of human disorders. Disregarding these limitations both in terms of modelling the initial insult (i.e. maternal obesity and maternal infection) and behavioural outcomes, preclinical studies have been instrumental in increasing our understanding of the pathophysiological mechanisms underlying associations between maternal inflammation and neuropsychiatric disorders in the offspring.
Despite some conflicting results, a common framework of behavioural alterations exists in each model. MIA is commonly used to model environment-induced impairments observed in schizophrenia and ASD. One of the most consistent outcomes in offspring of MIA is social deficits. Both Poly: IC and LPS administration during pregnancy lead to decreased communication and social interactions in mouse and rat models (Gumusoglu and Stevens, 2019). Similar findings were observed in mouse MHFD offspring (Buffington et al., 2016; Kang et al., 2014). Rodent MIA models also exhibit core behavioural phenotypes related to schizophrenia, including sensorimotor gating deficits, attention impairments, increased anxiety and cognitive impairments (Brown and Meyer, 2018; Gumusoglu and Stevens, 2019). Within this set of behavioural deficits only anxiety and cognition has been extensively studied in MHFD models, potentially because of the lower association between schizophrenia and maternal obesity in epidemiological studies (Jones et al., 1998). Several studies demonstrated increased anxiety-like behaviours in adults and juvenile MHFD offspring (Abuaish et al., 2018; Manti et al., 2018; Yan et al., 2017). The few conflicting results showing no effect or decreased anxiety might come from reduced exposure period and/or milder enrichment in dietary fat (40% vs 60% of kcal) (Balsevich et al., 2016; Hiramatsu et al., 2017). Cognitive defects are also commonly observed in MHFD offspring with impaired spatial learning and reduced learning rates (McKee et al., 2017; Park and Kim, 2017; Sanguinetti et al., 2019). In addition, these animals show increased motor activity and heightened hedonic behaviour (Balsevich et al., 2016; Fernandes et al., 2012; Hiramatsu et al., 2017; Peleg-Raibstein et al., 2016). Together, these studies suggest that MIA and MHFD impair common traits in offspring such as social, anxiety and cognition-related deficits, yet they might retain some specificities, for example, regarding complex behavioural impairments observed in schizophrenia. While consistent with epidemiological studies showing that both maternal obesity and maternal infection associate with higher risk for several neuropsychiatric disorders, standardised systematic studies are required to clearly establish similarities and differences between the two models.
Immunomodulation in pregnancy and obesity
During pregnancy, the immune system must ensure protection from external pathogens as well as prevent rejection of the semi-allogenic foetus. Research in the field has provided conflicting results in part owing to the use of animal models with species-specific differences in immunology and placental anatomy (Ander et al., 2019). Even comparison between human studies has limitations due to variation in study design, such as the period studied, cytokines measured or biological variation between ethnic groups (Gillespie et al., 2016; Graham et al., 2017). Nevertheless, consensus has moved from a predominantly immune-suppressant model to a more complex explanation. During implantation, when the blastocyst adheres to the uterine epithelium and begins to invade the endometrium, an inflammatory reaction, associated with an upregulation of interleukin-6 (IL-6) and tumour necrosis factor (TNF) among other cytokines (Griffith et al., 2017), allows the repair of the uterine epithelium following blastocyst invasion (Figure 1) (Yockey and Iwasaki, 2018). Post implantation, a preponderance of anti-inflammatory cytokines promotes tolerance of foetal antigens (Graham et al., 2017). Then, as parturition begins, an upregulation of pro-inflammatory signalling including IL-6 and TNF assists with the progression of labour (Rinaldi et al., 2017).
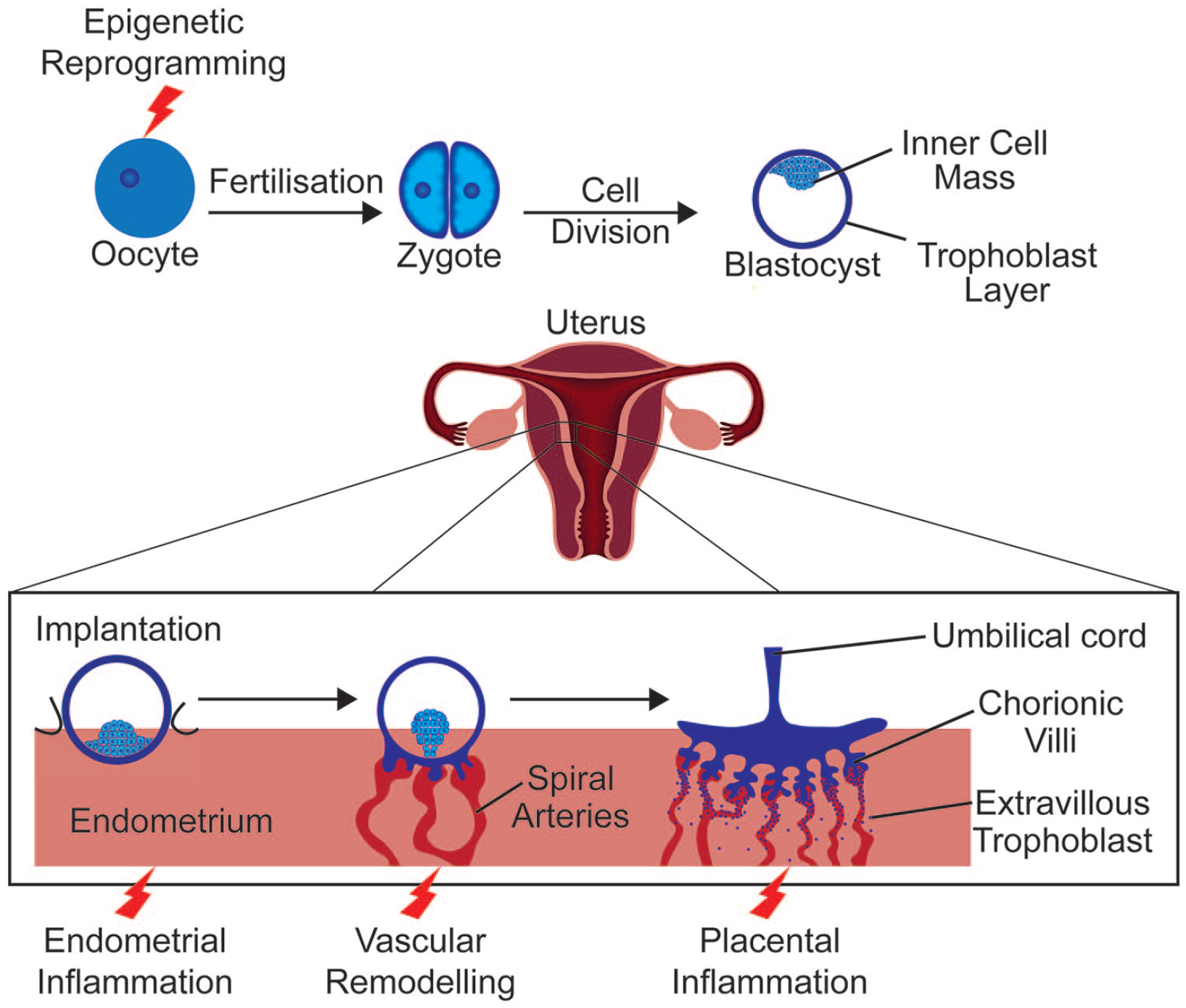
Early stages of pregnancy.
IL-6 and TNF-α are classical immune mediators involved in acute inflammation and are upregulated after infection in both humans and animal models (Glass et al., 2019; Holub et al., 2013). Other cytokines elevated after infection include C-reactive protein (CRP), predominantly after microbial infection in humans, although its role seems to be limited in mouse models (Holub et al., 2013; Torzewski et al., 2014). Evidence has also accumulated for the dysregulation of the immune system during obesity. Termed ‘meta-inflammation’ this inflammatory response is brought about by metabolic signals engaging inflammatory pathways in adipose tissues leading to increased immune cell infiltration. This results in increased pro-inflammatory cytokine production and further immune cell infiltration (Gregor and Hotamisligil, 2011; Weisberg et al., 2003). This meta-inflammation is chronic and low-grade and thus differs from the acute injury–induced or infection-induced inflammation (Gregor and Hotamisligil, 2011). Yet, these differing inflammatory conditions involve the very same cytokines. The study of obese rodent models first demonstrated elevated levels of TNF-α (Hotamisligil et al., 1993). Further studies in both animal models and humans identified other inflammatory mediators dysregulated by obesity, including IL-6, transforming growth factor β (TGF-β) and CRP (Khaodhiar et al., 2004; Samad et al., 1997).
During pregnancy, the impact of pathogen-induced immune activation on cytokine levels is largely the same as in non-gravid conditions, with increases in IL-6, TNF-α and CRP observed in both human and animal work (Fricke et al., 2018; Singh et al., 2018). The inflammatory state during maternal obesity is more controversial than during maternal infection. Animal literature is contradictory, with some papers finding increases in pro-inflammatory cytokines (Kim et al., 2014), while others find no change despite elevated foetal pro-inflammatory cytokines (Crew et al., 2016; Desai et al., 2013). Contrasting results could reflect inter-species variation or differences in diets as both high-fat and high-sugar/high-fat diets are used to induce obesity. As described for healthy pregnancy, human studies are complicated by study design limitations and biological variation. Three heavily measured cytokines, IL-6, TNF-α and CRP, report conflicting associations with body mass index (BMI) (Pendeloski et al., 2017; Sureshchandra et al., 2019). IL-6 is consistently higher in obese pregnant women than lean counterparts and concentration varies with gestation (Christian and Porter, 2014; Friis et al., 2013; Sen et al., 2014). An association between elevated CRP and maternal BMI is supported by most studies (Christian and Porter, 2014; Sen et al., 2014), yet a large longitudinal study did not corroborate this (Friis et al., 2013). Interestingly, levels of CRP closely correlated with gestational weight gain, suggesting a dose–response relationship, unlike IL-6 or TNF-α (Hrolfsdottir et al., 2016). The association of TNF-α with maternal obesity is least supported with many studies finding no correlation (Sen et al., 2014; Zembala-Szczerba et al., 2017). These findings highlight the complexity of the inflammatory state when obesity and pregnancy interact and point towards some level of immune dysregulation during maternal obesity.
Effects of obesity on placental inflammation
Obesity is a risk factor for pregnancy complications such as preeclampsia and adverse foetal outcomes like preterm birth (Papachatzi et al., 2013; Sohlberg et al., 2012; Spradley et al., 2015). The materno-foetal interface of the placenta plays a crucial role in pregnancy success by setting the intrauterine environment for the foetus. A recent genome-wide association study highlighted that genetic risk for schizophrenia is higher in patients with history of early life complications (Ursini et al., 2018). The genes conveying this early life complications–associated genetic risk converge on hypoxic stress, metabolism and immune function pathways in the placenta suggesting they might be key in ‘malprogramming’ the brain for later dysfunction.
Interestingly, placentae from obese mothers or after episodes of maternal infection show higher signs of hypoxic stress and impaired vasculature (Gohir et al., 2019; Kalk et al., 2017; Moeller et al., 2019). In MHFD mouse models, the number of foetal endothelial cells in the placenta is reduced (Kretschmer et al., 2020); placental blood vessels are dysfunctional and lack pericytes (Gohir et al., 2019). Accordingly, transcriptomic studies comparing placental tissue from lean and obese women identified modules of differentially expressed genes related to immune function and blood vessel formation, suggesting defective vascularisation and immunity are critical impairments in obesity-induced placental defects (Cox et al., 2019).
Immunomodulation is essential for the development of the placenta, which relies on complex interactions between the foetal tissue and the uterine mucosa. Recently, complex ligand–receptor pairs controlling interactions between maternal immune cells and invading foetal trophoblast cells were identified (Vento-Tormo et al., 2018). Tissue-resident (decidual) natural killer cells (dNK) interact with trophoblasts and are thought to promote their migration and recruitment through chemokine signalling. In return, invading trophoblasts express immunomodulatory molecules that bind dNK’s killer immunoglobulin-like receptors (KIR), preventing inflammatory reactions. Acute and low-grade inflammation associated with infection and obesity, respectively, likely perturbs this highly orchestrated process. Indeed, MIA leads to an acute increase in activated decidual immune cells, including dNK (Hsiao and Patterson, 2011) and maternal obesity increased the number of activated dNK in the placenta both in human and mouse studies (Baltayeva et al., 2020; Castellana et al., 2018). Intriguingly, only subtle differences in transcriptional programmes were detected between obese and lean dNKs, possibly due to a lack of cellular resolution. It would be interesting to revisit this issue in light of the most recent single-cell advances in the field.
dNK are not the only immune cells affected by maternal obesity in the placenta. A large study on placentae of 400 pregnant women indicates that obesity associates with chronic villitis (Leon-Garcia et al., 2016), an inflammation of the chorionic villi that provide essential exchanges between maternal and foetal blood. Both CD3+ T cells and CD68+ macrophages infiltrated the villi, likely secondary to the initial inflammation. Interestingly, chronic villitis is a risk factor for cerebral palsy and neonatal encephalopathy (Chen and Roberts, 2018; Redline, 2005), highlighting the link between placental inflammation and insults to the developing brain. The scenario leading to placental inflammation is unclear, but a tentative hypothesis is that initial defects in communication between dNK and trophoblasts perturb proper development of the materno-foetal interface, including blood vessels, which would trigger hypoxia and placental inflammatory events. While the placenta itself produces cytokines that might enter foetal circulation and impair brain development, it is not clear how maternal obesity affects this process.
Maternal obesity and the role of cytokines in neural development
In addition to their roles in pregnancy, cytokines are involved in brain development. IL-6 and members of TGF and TNF families regulate many developmental processes from initial central nervous system (CNS) formation to synaptogenesis (Deverman and Patterson, 2009). Studies examining associations between cytokine genetic polymorphisms and neuropsychiatric disorders suggest a link between higher levels and increased risk for these disorders. The TNF-α variant -238 G>A, for example, which may increase circulating TNF-α levels, was associated with schizophrenia in a Tunisian population (Inoubli et al., 2018) but not in Indian Bengalee patients (Debnath et al., 2013). Studies on genetic variation in IL-6 have provided similarly conflicting results. However, a recent study identified a single nucleotide polymorphism (rs2228145) influencing IL-6 receptor concentrations, as being significantly elevated in French early-onset bipolar disorder patients and trending to increase in Indian Tamil patients. This suggests a trans-ethnic association between bipolar disorder and this polymorphism (Sundaresh et al., 2019). Given the importance of these signalling molecules in development, one could hypothesise that their dysregulation through maternal obesity may contribute to increased neurodevelopmental disorder susceptibility in offspring.
IL-6 belongs to the neuropoietic cytokine family which regulates self-renewal of neuroepithelial cells (Deverman and Patterson, 2009). IL-6 also regulates axon development by reducing neurite outgrowth of cultured hypothalamic tissue. Interestingly, circulating IL-6 is increased in the newborn offspring of obese mice and reduced innervation of the hypothalamic paraventricular nucleus in these animals suggests dysregulated cytokine levels in offspring of obese mothers may impair formation of hypothalamic circuits (Sanders et al., 2014). TNF-α is another cytokine shown to be involved in neurite outgrowth. Soluble TNF-α reduced neurite outgrowth during a specific period of late embryonic and early postnatal development (Nolan et al., 2014). Reductions in axon growth were also observed in neuronal populations within the sympathetic nervous system upon TNF exposure (Erice et al., 2019). These findings raise the possibility that increased TNF-α expression resulting from maternal obesity could affect neurite growth, at least within the sympathetic nervous system.
TGF-β signalling is involved in much of early CNS development (Meyers and Kessler, 2017). Dopaminergic neurons, for example, require TGF-β signalling for differentiation, maintenance, axon growth and regulation of synaptic electrophysiology (Farkas et al., 2003; Luo et al., 2016). TGF-β was also recently shown to regulate dendritic spine morphology in midbrain neurons (Yoon et al., 2020). Interestingly, a common TGFB1 polymorphism, correlated with increased TGF-β production, was associated with increased schizophrenia susceptibility (Frydecka et al., 2013). This is corroborated by elevated TGF-β expression observed in circulating monocytes of treatment-free schizophrenia patients (Amoli et al., 2019). Decreases in hippocampal TGF-β expression were found in male offspring of MIA rats with repetitive behaviours and reduced measures of social interaction associated with ASD (Fortunato et al., 2017). Decreased TGF-β levels have also been identified in children with ASD (El Gohary et al., 2015). This suggests a fine balance of TGF-β levels is required for typical neurodevelopment, a balance that can be affected by obesity. Importantly, the brain’s resident immune cells, microglia, require TGF-β signalling for development and function (Butovsky et al., 2014; Lively et al., 2018). Modulation of microglia represents another mechanism by which inflammatory cytokines could impact brain development. Most research into the effect of cytokines on neurons has been conducted in adult models. While the impact of viral infection on foetal neurodevelopment through interferons is better understood, more research is required on how other cytokines affected by maternal obesity directly influence neurodevelopment.
The impact of maternal obesity on microglia
Microglia are heavily involved in brain development, as covered recently by many excellent reviews (Hammond et al., 2018; Li and Barres, 2018; Thion et al., 2018a). Microglia have also been implicated in the pathogenesis of ASD, schizophrenia and major depressive disorder among other conditions (Tay et al., 2017). Their presence from the beginnings of brain formation and ability to sense the environment place them at a key position to mediate the effects of maternal obesity. As resident immune cells of the CNS, microglia must react to signs of pathogen invasion. Pathogen-associated molecular patterns like LPS trigger acute systemic and CNS inflammation. While vital for the removal of pathogens, immune activation can interfere with the developmental responsibilities of microglia, as covered in Box 1.

Microglia’s developmental roles and how they are affected by maternal immune activation.
Maternal obesity can prime microglia towards a pro-inflammatory phenotype postnatally, as demonstrated in rats exposed to maternal obesity with increased levels of microglial activation marker CD11b within the hippocampus at birth (Bilbo and Tsang, 2010). Subsequent LPS stimulus induced exaggerated hippocampal microglia activation and increased IL-1β production. These defects were accompanied by increased anxious behaviour among male offspring. Maldonado-Ruiz et al. (2019a) demonstrated that maternal obesity-exposed adult hypothalamic microglia become increasingly activated following grehlin-mediated neuron activation. Less is known about the impact of maternal obesity on foetal microglial populations. One key study found that while maternal obesity did not induce a pro-inflammatory state, foetal microglia were ‘primed’ to produce more TNF-α upon LPS stimulation than controls (Edlow et al., 2019). Interestingly, placental macrophage populations strongly correlated with foetal microglia in LPS-stimulated TNF-α production. Prenatal priming also occurs in MIA (Mattei et al., 2017). This may increase susceptibility to neuropsychiatric disorder through interactions with insults later in life. Interactions between MIA and later life psychological trauma do appear to synergistically increase risk for schizophrenia in males (Debost et al., 2017).
Several mechanisms may contribute to the impact of maternal obesity on microglial biology. For example, the adipose-derived hormone leptin regulates microglial activation state in the rat hypothalamus and induces inflammatory cytokine production in vitro (Gao et al., 2014). Leptin receptor deficiency in myeloid cells, generated by Cre-Lox recombination, impairs microglial phagocytosis and decreased mouse hypothalamic neuron number (Gao et al., 2018). Interestingly, leptin circulating levels are increased in the offspring of obese dams (Chen et al., 2008). This suggests that leptin signalling might regulate microglia phenotype in the developing brain of obese offspring.
Another example is the role of microglia metabolism. Milanova et al. (2019) show that microglia, but not circulating myeloid cells, in obese rats increase lipid metabolism–associated gene expression and decrease expression of glycolytic genes. Metabolic reprogramming plays a key role in regulating macrophage phenotype and plasticity. The initial view of pro-inflammatory glucose metabolism versus anti-inflammatory fatty acid oxidation has moved to a more complex picture of metabolic flexibility driving macrophage states (Van den Bossche et al., 2017). Recent studies started to examine this phenomenon specifically in microglia. Pro-inflammatory microglia have increased glucose metabolism that preferentially fuels the tricarboxylic acid (TCA) cycle, which is also fuelled by increased glutamine oxidation. Indeed, glutamine is used as an alternate carbon source upon acute hypoglycaemia to maintain microglia surveillance function (Bernier et al., 2020). Both mitochondrial fatty acid oxidation and fatty acid synthesis are higher in anti-inflammatory polarised microglia and decreased in inflammatory states. On the other hand, lipid droplets are frequently found upon inflammation in immune cells and in microglia after LPS stimulation or in aging brain (Marschallinger et al., 2020). These lipid droplet–containing microglia have impaired phagocytic function and a pro-inflammatory secretome. Metabolic disruptions during maternal obesity might therefore impact microglial metabolic plasticity in the offspring that would in turn impair neurodevelopment.
The immunomodulatory roles of fatty acids
In addition to increased levels of inflammatory cytokines, obese women show an altered profile of circulating metabolites, including fatty acids (Tu et al., 2019). Among these, saturated fatty acids (SFAs) and polyunsaturated fatty acids (PUFAs) and their derivatives have the best characterised immunomodulatory properties.
SFA gavage increases microglial expression of activation markers, including TNF-α, without increasing peripheral inflammatory cytokine levels in adult mice (Valdearcos et al., 2014). Moreover, microglial depletion reduced hypothalamic inflammation and neuronal stress marker Hsp72 demonstrating microglia are responsible for SFA-mediated hypothalamic neuronal stress. As microglia are brain-resident macrophages, the mechanisms of SFA-induced activation are likely similar to those in other macrophage populations. SFAs were thought to cause macrophage activation as agonists of Toll-like receptor 4. However, Lancaster et al. (2018) provide compelling evidence that this effect may be mediated by changes in macrophage metabolism. Recent evidence suggests fatty acid metabolism is involved in regulating activation through modulation of plasma membrane properties, rather than directly inducing polarisation or binding Toll-like receptors (Divakaruni et al., 2018; Köberlin et al., 2015). As SFAs are elevated in obese versus lean pregnancies (Vidakovic et al., 2015), these bioactive lipids may contribute to developmental inflammation. SFAs have been shown to induce pro-inflammatory cytokine release in placental trophoblast cells which, as mentioned previously, may contribute towards neurodevelopmental disorder (Yang et al., 2015). Yet, evidence for the transport of SFAs across the placenta is limited, with human studies finding no correlation between circulating maternal and foetal SFA levels (Liu et al., 2017; Meher et al., 2016). Further studies should determine whether and how SFA can cross the placental barrier and how they affect inflammatory processes in the foetus.
The major source of PUFAs in mammals comes from the diet as precursors found in vegetable oils or directly from meat (n-6) and fish (n-3). In a schematic view, n-6 PUFAs are converted into eicosanoids and prostaglandins with pro-inflammatory properties. By contrast, n-3 (also termed omega-3) PUFAs have anti-inflammatory effects through conversion into specialised pro-resolving mediators such as resolvins (Rey et al., 2019). Importantly, maternal dietary manipulations of PUFA content induce modifications in the lipid profile of the developing brain. An imbalanced diet with high levels of n-6 and low levels of n-3 PUFAs results in corresponding changes in the foetal brain, without affecting global levels of saturated or monounsaturated fatty acids (Sakayori et al., 2016). Maternal dietary manipulations alter PUFA content of microglia in the offspring brain with high n-6/n-3 PUFA ratio leading to a corresponding increase in n-6 PUFAs in microglia membrane. Conversely, n-3 PUFA supplemented diet with low n-6/n-3 PUFA ratio resulted in higher n-3 PUFA membrane content. Intriguingly, n-3 PUFA supplemented diet had a stronger effect on microglia lipid composition, including phospholipid classes (Rey et al., 2018). These changes might be linked to impaired microglia function, as maternal dietary n-3 PUFA deficiency impaired microglial process motility and regulated expression of inflammation-related genes (Madore et al., 2014). In addition, n-3 PUFA deficiency exacerbated inflammation and behavioural defects induced by MIA (Labrousse et al., 2018). While these studies clearly show the adverse effect of maternal dietary PUFA deficiency on prenatal brain development and behavioural outcome in offspring, our understanding of the contribution of PUFAs to foetal development and cognitive outcome in maternal obesity is much more limited.
MHFD models rely on diets usually enriched in SFAs with a high n-6/n-3 PUFA ratio, reflecting the Western diet. Indeed, obese pregnant women consistently have an elevated n-6/n-3 PUFA ratio (Benaim et al., 2018; Vidakovic et al., 2015). Interestingly, a study in Hawaiian pregnant women, whose fish consumption is likely higher than in the typical Western diet, showed that obese women had higher n-3 PUFA levels than obese women from Ohio, albeit lower than their lean counterparts (Alvarado et al., 2018). Placenta from Hawaiian obese women did not show signs of lipid accumulation and had similar levels of TNF-α, suggesting that higher intake of n-3 PUFAs might have a protective effect in the placenta. Interestingly, dietary PUFAs also seem to have a protective effect against changes in the epigenetic landscape of the placenta. Female rats fed a high-fat diet enriched in SFAs had increased methylation but reduced histone acetylation in the placenta, compared to those fed a diet enriched in long-chain or very-long-chain PUFAs (Ramaiyan and Talahalli, 2018). Human studies of maternal n-3 PUFA supplementation yielded controversial results regarding beneficial effects on offspring cognitive outcome (Colombo et al., 2019; Ramakrishnan et al., 2016). One reason for this might be that beneficial effects are more likely to be observed in adverse conditions such as maternal obesity. Maternal dietary n-3 PUFA deficiency and maternal obesity lead to similar defects in prenatal brain development and behavioural impairments in animal models. Hence, they might share common pathways worth investigating further to provide new leads for intervention.
Maternal obesity alters the epigenetic landscape in offspring
Cellular metabolism is tightly linked to epigenetic modifications. Lipid pathways provide acetyl-coA and β-hydroxybutyrate which are involved in acetylation/deacetylation reactions and the mitochondrial TCA cycle also provides molecular substrate for DNA methylation. Thus, altered metabolism in the context of obesity might affect epigenetic processes throughout foetal development.
Shortly after fertilisation, a global demethylation accompanied by oxidation of 5-methylcytosine (producing 5hmC marks) contributes to resetting the paternal genome. By contrast, the maternal pronucleus is protected from active demethylation early on by the DNA-binding protein Stella and demethylation is achieved by dilution in subsequent divisions (Lee et al., 2014; Smith and Meissner, 2013). Han et al. (2018) show in an elegant study that this epigenetic remodelling is altered under MHFD. The epigenetic asymmetry usually observed between maternal and paternal genomes is lost in MHFD zygotes, with increased hypomethylation across the genome in particular at transposon elements. They further show that Stella levels are decreased in MHFD oocytes and restoring Stella levels prevents the early loss of epigenetic asymmetry contributing to the maintenance of the early embryonic methylome (Han et al., 2018). Thus, defects in the maternal epigenetic machinery due to diet-induced obesity have long-term consequences on the epigenetic programme required for proper embryonic development.
Strikingly, a global increase in methylation with concurrent decrease in hydroxymethylation of DNA across the genome is observed in placenta from obese women (Mitsuya et al., 2017; Ramaiyan and Talahalli, 2018), potentially regulating immune response, placental and foetal development gene programmes. A recent study showed that epigenetic modifications also occur in the immune cell lineage during development. Umbilical cord blood monocytes from babies of mothers with pregravid obesity had decreased methylation in promoter regions of genes involved in inflammatory response. These changes in the epigenetic profile of umbilical monocytes did not affect their transcriptional programme at resting state but blunted their response to LPS stimulation and the associated transcriptional changes (Sureshchandra et al., 2019).
How these global alterations specifically affect inflammation in the developing brain is unclear. Dynamic changes in the epigenome are a major feature of proper microglia development. The landscape of accessible chromatin regions is strikingly different between embryonic and adult microglia (Matcovitch-Natan et al., 2016). Genetic deletion prenatally of two histone deacetylase genes Hdac1 and Hdac2 impaired microglia development with long-lasting effects later in life, contrasting with little effect when performed postnatally (Datta et al., 2018). Moreover, the unique signature of microglia heavily relies on environment-dependent epigenetic regulation, which critically is lost when they are placed in culture (Gosselin et al., 2017). Importantly, the genes downregulated in cultured microglia largely overlap with those upregulated during in vivo development, indicating that microglial developmental programmes require interaction with the environment. Indeed, the epigenetic landscape of embryonic microglia is sensitive to environmental changes such as absent maternal microbiota (Thion et al., 2018b). Given the widespread alterations in DNA methylation upon exposure to maternal obesity and the tight link between DNA methylation and histone modifications, it is likely that microglia’s epigenetic landscape is impaired in offspring of obese mothers. Surprisingly, while MIA induces changes in embryonic microglia’s transcriptome, modifications in their epigenetic programme have not been reported. These MIA-induced transcriptomic changes seem to be transient as adult microglia show only mild differences in gene expression (Matcovitch-Natan et al., 2016; Mattei et al., 2017).
A role for extracellular vesicles in mediating the impact of maternal obesity on the developing brain?
The question of how dysregulated signals from obese mothers reach the developing brain is a complex unanswered issue. Circulating cytokines and nutrients might enter foetal circulation after tight control at the level of the placenta, but this process is likely compound specific and our understanding is still limited. Another layer of complexity comes from extracellular vesicles (EVs). EVs describe a broad range of cell-released membranous structures. EV nomenclature is ambiguous with the term exosome used to describe EV origin or size – usually 30–100 nm for exosomes. To address this, EV nomenclature guidelines have recently been produced by experts in the field (Théry et al., 2018). Here, the term exosome is used only where authors have experimentally established endosomal origin of the EV. EVs are involved in intercellular signalling during many processes, including embryonic and neurodevelopment (Gong et al., 2016; Greening et al., 2016). Signalling occurs through transport of RNAs, DNA, proteins or bioactive lipids from donor to recipient cells or through ligand–receptor interactions at the EV and cell surface (Iraci et al., 2016). Exosomes are a specific type of EV of endosomal origin that have received much attention for their roles in cell–cell signalling. Importantly, exosomes can cross both the placental (Sheller-Miller et al., 2019) and blood–brain barriers when injected systemically (Alvarez-Erviti et al., 2011).
During brain development, EVs are released by neurons, glia, oligodendrocytes and astrocytes. These EVs are involved in several processes including trophic support and inflammation (Sharma et al., 2013). Human macrophage EVs, for example, increase pro-inflammatory cytokine release in placental trophoblast explants (Holder et al., 2016). EVs may therefore contribute towards placental inflammation during maternal infection leading to neurodevelopmental disorder as mentioned earlier. The transfer of Zika virus between murine cortical neurons has been shown to occur through EVs and some studies indicate trans-placental transport of Zika virus containing EVs (Zhang et al., 2016; Zhou et al., 2019). In addition to its direct effect on neural progenitor survival, Zika infection of cranial neural crest cells increased cytokine production in vitro leading to altered neurogenesis and apoptosis of neural progenitors (Bayless et al., 2016).
Developmentally EVs transport morphogens such as Wnt and Sonic Hedgehog (Shh), which are instrumental in vertebrate brain development influencing cell proliferation, patterning and differentiation (Memi et al., 2018). Shh-containing exosomes are released by chick embryo notochord cells and human cell lines, initiating differentiation of mouse embryonic stem cells into motor neurons (Vyas et al., 2014). Interestingly, exosomal Shh released by adipocytes induces pro-inflammatory polarisation of macrophages and maternal obesity increases human circulating pro-inflammatory exosome number (Elfeky et al., 2017; Song et al., 2018). Expression of Shh is also increased in the subcutaneous fat of obese mice (Yao et al., 2019). Combined, these data present the possibility that maternal obesity–induced changes in Shh exosomes impact foetal neurodevelopment by directly modifying neural Shh signalling or by inducing a pro-inflammatory state in brain macrophages.
Adipose tissue may be an important source of exosomal signalling, including via micro-RNAs (miRNAs) (Thomou et al., 2017). Exosomes from obese individuals contained 55 differentially expressed miRNAs, relative to lean counterparts, some of which target TGF-β signalling (Ferrante et al., 2015). Obesity-induced changes to exosome profile have a functional impact on macrophages. Increased expression of miRNA-34a in mouse adipose tissue exosomes enhanced pro-inflammatory polarisation of adipose macrophages (Pan et al., 2019), another potential mechanism by which brain macrophage behaviour could be altered. Furthermore, adipocytes may have an exosome-mediated effect on macrophage differentiation during obesity (Flaherty et al., 2019). Microglia differentiation in particular is susceptible to TGF-β signalling modification which may be impacted by exosomes in obesity (Ferrante et al., 2015; Utz et al., 2020).
Findings summarised here suggest exosome signalling during obesity causes changes to macrophage behaviour and inflammatory status. In maternal obesity, these changes could be mirrored in the foetal brain resulting in neurodevelopmental abnormalities (Figure 2). Indeed, modulation of maternal exosome–derived miRNAs through metabolic dysfunction is already known to induce developmental defects in other systems (Shi et al., 2017). It has also been proposed that hyperlipidaemia might cause release of mitochondrial DNA–containing EVs (Maldonado-Ruiz et al., 2019b). Serum of children with ASD is known to contain EVs with elevated mitochondrial DNA able to induce microglial pro-inflammatory cytokine expression (Tsilioni and Theoharides, 2018). Further investigations are needed to better understand the role of exosomes in transmitting signals of maternal obesity to the developing brain.

Proposed mechanism for exosome-mediated microglia activation in maternal obesity.
Future directions and intervention strategies
A number of inflammation-related systems are dysregulated under obesity, systems with a major role in successful offspring development. This is evident from observed similarities in neuropsychiatric outcomes between MIA and maternal obesity. Several hurdles lie in the path to fully understanding the mechanisms involved in the inflammatory impact of maternal obesity on neurodevelopment. Clearing up contradictory findings in cytokine circulation under maternal obesity would be an excellent start. Longitudinal studies measuring cytokine levels in obese women at regular intervals throughout pregnancy and afterwards, as carried out by Graham et al. (2017) for healthy pregnancy, could begin to clarify understanding provided ethnic differences are accounted for. Differences in diet between subjects may also need to be controlled for given the modulatory effects of n-3/n-6 PUFAs on inflammation (Kanerva et al., 2014; Shivappa et al., 2018). In addition, measuring cytokine levels in placental tissue and cord blood under these conditions could shed light on the relationship between maternal, placental and foetal cytokine profiles during maternal obesity. Knowledge in this field may allow better targeting of any anti-inflammatory therapies to particular stages of pregnancy or women with particular complications.
Further research is also required within the field of microglial biology to understand the role of leptin signalling and metabolic dysfunction in the developmental activity of microglia. Further investigation into dietary modifications with a focus on microglia could provide intervention opportunities. Low carbohydrate ketogenic diet metabolites, for example, reduce microglial inflammation but are likely not appropriate during pregnancy (Huang et al., 2018). Studies examining changes in microglial metabolism under maternal obesity could also help uncover epigenetic changes in microglia, given the close relationship between the two. In addition, investigating the crosstalk between microglia and other cells that contribute to brain inflammation including astrocytes will shed light on cellular interactions critical for sustained effect on brain functions.
Initial studies into the role of exosome signalling in microglia behaviour are also required. The potential for exosomes in the treatment of many disorders has begun to be explored. Preclinical investigations suggest these vesicles may be useful in the treatment of ASD (Perets et al., 2018). Although great care is needed when applying preventive treatments to pregnant women, the fact that exosomes that are naturally involved in pregnancy may reduce the risk of complications if applied to maternal obesity. The targeting of inflamed regions, including during neuroinflammation, by certain exosomes makes anti-inflammatory exosome treatment an interesting avenue of investigation (Perets et al., 2019).
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: J.D was supported by a Hodge Foundation PhD studentship. E.M. is a recipient of the Hodge Foundation Fellowship and received support from a Springboard Award from the Academy of Medical Sciences (SBF005\1100).