
Abstract
This review provides a distillate of the advances in knowledge about the neurotransmitter functions of acetylcholine over the 50-year period between 1967 and 2017, together with incremental information about the cognate nicotinic and muscarinic acetylcholine receptors, and some brief comments on possible advances in the near future. The text is supplemented by a timelines figure indicating the dates of some key advances in knowledge about acetylcholine receptors and a box-figure providing a snapshot of selected papers about acetylcholine published in the year 1967.
Keywords
Introduction: the research environment 50 years ago
In order to appreciate what was known or not known 50 years ago, and if not, why not, it is important to know what currently used facilities were not available to the lab neuroscientist pre-1967.
Thus, although mainframe computers (accessed by punched cards) were coming in there were no PCs or lab computers. (The best our lab could afford in 1967 was a 64-step programmable calculator costing more than a PC does now.)
Although the structure of DNA was known (in 1953) and the genetic code had been unravelled in 1962, there were no ways of gene-cloning or gene manipulation, and no ‘knock-out’ mice or knock-down siRNA to test what a gene did.
There was also neither chemical nor structural information about membrane proteins such as receptors and ion channels, and no means of seeing their location with antibodies or mRNA hybridisation.
The electrophysiologist was restricted to using microelectrodes (no patch-clamp) for recording and drug application, with no visual aids for seeing neurons like GFP, no calcium indicators for monitoring activity or optogenetics for tracking circuits.
When it came to recording data, this was usually done directly on photographic film (subject to the hazard of dark-room development, when all was lost if someone switched the light on) – neither computer corrections nor enhancement was available.
Papers were written on a typewriter (with carbon paper copies – no word processors or photocopiers), or sometimes just by hand, and submitted for publication by post. There were no e-mails or Internet, and no electronic journals – to read the references meant going to the library with a notebook or a bunch of index cards in hand.
Under the circumstances, one can only be impressed by how much was discovered
What was known by 1967
Acetylcholine as a neurotransmitter
By 1967, acetylcholine (ACh) was firmly accepted as a major neurotransmitter in the peripheral nervous system, including somatic motor nerves and parts of the autonomic nervous system (see, e.g. Goodman and Gilmam, 1965; Krnjevic, 1974). Enzymes for its synthesis ( ‘choline acetylase’ = choline acetyltransferase) and degradation (cholinesterase) had been isolated and studied biochemically. ACh release following nerve stimulation had been detected from vagal parasympathetic nerves, preganglionic sympathetic nerves, cholinergic postganglionic sympathetic nerves and somatic motor nerves. A transmitter function was also supported by inhibition by tubocurarine (motor nerves, preganglionic sympathetic nerves) or atropine (postganglionic parasympathetic nerves).
Transmission at the neuromuscular junction
Comprehensive information regarding the details of somatic nerve-to-muscle transmission had been generated by the work of Bernard Katz and his colleagues (Katz, 1966). Fatt and Katz (1951) used R.W. Gerard’s recently introduced microelectrode technique (Ling and Gerard, 1949: J. cell. comp. Physiol, 34,383 383) to make the first intracellular recordings of the end-plate potential from the frog neuromuscular junction. Using the muscle action potential as a neat way of altering membrane voltage, they deduced that the epp arose from a general increase in ionic conductance (cations and anions) which partially short-circuited the action potential. On the basis of further studies with radioactive tracers (Jenkinson and Nicholls, 1961) and reversal potential measurements under voltage-clamp (Takeuchi and Takeuchi, 1960) the conductance change was adduced to be only to the cations Na+ and K+, not anions. This became the model for other forms of excitatory synaptic transmission (Eccles, 1957; Ginsborg, 1967): Apart from the facts that they could be activated by acetylcholine and nicotine (and hence was classified as ‘nicotinic’ following Dale’s (1914) nomenclature), and inhibited by tubocurarine and related alkaloids, the physical nature of the muscle end-plate receptor was entirely unknown. [Fatt and Katz (1951) did not even mention a receptor – they only referred to an interaction of acetylcholine with the end-plate membrane.] One approach to the receptor was to use ligand binding to find out more about it. Thus, Peter Waser (1960) used radioactively-labelled tubocurarine to begin to localise the end-plate receptors by autoradiography. However, the resolution was poor and microscopic resolution had to await the later introduction of α-bungarotoxin. Also, Waser and others (e.g., Chothia, 1970) tried to deduce the chemical nature of the acetylcholine binding site from studies comparing chemical congeners.
Transmitter release
Another crucial advance from Katz’ work on the frog neuromuscular junction was the discovery of miniature epps (Fatt and Katz, 1952), which led to the development of the quantal theory of transmitter release (see Katz, 1969). This, coupled with the discovery of synaptic vesicles (de Robertis and Bennett, 1955), provided the foundation stones for nearly all subsequent studies on transmitter release at synapses.
Transmission between neurons
Pre-1967, intracellular microelectrode recordings were also obtained from sympathetic neurons, another prospective site of tubocurarine-sensitive nicotinic cholinergic transmission (Blackman et al., 1963a, 1963b; Eccles, 1955; Nishi and Koketsu, 1960). These revealed a very similar transmission process to that at frog muscle end-plates – a depolarising excitatory postsynaptic potential (epsp), giving rise to a superimposed action potential; and spontaneous mepsps (though at a low frequency unless enhanced by raising (K+)out) forming the quantal components of the epsp: Notwithstanding, other experiments using extracellular recording methods, were beginning to suggest the presence of slower synaptic processes following repetitive afferent stimulation that were mediated by muscarinic (atropine-sensitive) receptors (see Phillis, 1970). The presence of a slower muscarinic component to the cholinergic excitation of Renshaw cells in the spinal cord (see below) was also emerging (Curtis and Ryall, 1966). Slow muscarinic excitatory effects were subjected to intensive study in subsequent years, generating new concepts of neural information processing and intracellular signalling mechanisms (see Brown, 2010). Another difference between the motor end-plate and the sympathetic ganglion already apparent by 1967 concerned the nature of the nicotinic receptors. Although both are sensitive to tubocurarine, in an attempt to control essential hypertension a number of selective ganglion-blocking drugs had been developed which had little effect on muscle receptors. These included hexamethonium (Paton and Zaimis, 1949), pentolinium (Mason and Wien, 1955), and mecamylamine (Stone et al., 1956). Much later (following the cloning of the nicotinic receptors it transpired that this difference between nerve and muscle receptors was related to their different subunit compositions (see later).
Transmission in the CNS
By 1967, there was plenty of evidence suggesting an important role for ACh in the CNS (see Feldberg, 1954; Krnjevic, 1974; Phillis, 1970). It was present therein in high concentrations, as were choline acetyltransferase and cholinesterase (Hebb, 1957). Using a histochemical assay, Shute and Lewis (1963 & elsewhere) describe specific aggregations of neurons and specific neural projection tracts containing a high concentration of acetylcholinesterase, suggesting that they were cholinergic (a designation later supported by co-localization with choline acetyltransferase: Levey et al., 1983). There was also pharmacological evidence for likely transmitter functions (Goodman and Gilman, 1965). Thus, injecting ACh itself into the brain via the cerebral ventricles produced a variety of behavioural effects. CNS-penetrant anti-cholinesterases (including the nerve gases DFP, sarin and tabun, developed during WWII) exerted a variety of central excitatory effects, plausibly caused by enhanced effects of naturally released ACh because they could be diminished by atropine. Nicotine also clearly had central effects, including inhibition of ADH secretion via the hypothalamus (replicated by local ACh injection). The lipophilic muscarinic agonists pilocarpine, muscarine and arecoline produced cortical EEG arousal, whereas hyoscine (scopolamine) desynchronised the EEG and inhibited the arousal effect of photostimulation or reticular formation activation; and scopolamine exerted a well-known amnesic effect (witness its use in obstetrics or pre-anaesthetic medication to produce ‘twilight sleep’). Atropine and scopolamine were also known to be effective in diminishing the tremors of Parkinson’s disease, and, with more lipophilic derivatives thereof such as benztropine, were the mainstay of Parkinson’s disease treatment until the advent of levodopa. Finally, the release of ACh from the surface of the cerebral cortex, and its enhancement by afferent stimulation, could be detected (Mitchell, 1963; see also Box 1).
A snapshot of 1967.

What about cholinergic Although analogous to cholinergic transmission at the neuromuscular junction and at autonomic ganglia, other and subsequent studies revealed some differences. First, like sympathetic neurons, Renshaw cells also possess excitatory muscarinic receptors (Phillis, 1970), though how far they contribute to cholinergic synaptic excitation seems unclear. Second, the co-release of glutamate with the acetylcholine also contributes to transmission between motor axon collaterals and Renshaw cells (Lamotte d’Incamps and Ascher, 2008) though not apparently to synaptic transmission at the peripheral end of the motor fibres onto skeletal muscle (Nishimaru et al, 2005) (Co-release of two transmitters was unheard of in 1967, but co-release of glutamate with acetylcholine from other “cholinergic” neurons in the CNS such as basal forebrain neurons (Allen et al., 2006: J Neurosci 26: 1588–1595) has since been reported (see also Lamotte d’Incamps and Ascher, 2008, for some more examples).. Contrary to common belief, it does not contradict “Dale’s Principal “ (that the same chemical should be released from all processes of the same neuron; Dale, 1935: Proc.Roy.Soc.Med., 28: 319–332) since Dale did not specify only one transmitter. However, the apparent selective release of glutamate from only the collateral terminals would seem to do so.) Fast excitatory cholinergic transmission has been identified at a few other synapses in the brain (see Lamotte d’Incamps and Ascher, 2008, for examples) but these are rare. Most nicotinic receptors in the brain seem to be presynaptic and most postsynaptic cholinergic effects are mediated by muscarinic receptors. (see Brown, 2010)
Advances 1967–2017
Nicotinic receptors
Individual receptor currents
Membrane ‘noise’ was recorded during ACh depolarisation of frog muscle end-plates using focal extracellular recording (Katz and Miledi, 1972): ‘… the orders of magnitude of the calculated “shot effect”… provide a basis for discussing certain questions which seemed previously not to be open to experimental attack. Among these are: the number of ionic gates involved in the production of a miniature e.p.p.; the absolute conductance of a single ion gate opened by ACh molecules; the duration of the gating action and the total transfer of charge through the ion channel; the relation between the time course of the elementary current and the kinetics of drug/receptor action; the probability of single or repeated action of individual ACh molecules during normal transmission, etc’. ACh-induced current fluctuations were subsequently recorded under voltage-clamp by Anderson and Stevens (1973: J.Physiol., 235: 655–691).
Single channel currents of ACh receptors were recorded from denervated skeletal muscle membranes (Neher and Sakmann, 1976): ‘Recordings of single-channel currents finally resolves the third level of quantitation in the process of neuromuscular transmission after the discovery of endplate currents and miniature endplate currents’. Resolution was improved with the introduction of the gigaseal patch (Hamill et al., 1981: Pflug.Arch., 391, 85–100). This allowed the molecular interaction of ACh molecules with single nicotinic receptors to be examined at high temporal resolution to obtain realistic rate constants for suggested kinetic schemes of agonist–receptor interaction (Colquhoun and Sakmann, 1985) and the basis for such mysterious concepts as ‘partial agonism’ to be determined (Lape et al., 2008: Nature, 454, 722–727; Figure 1).

Cholinergic receptors: discovery timeline 1967–2017.
Clones and genes
Using the electric organ (electroplax) of the electric eel Torpedo as a rich source of muscle-type nicotinic receptors, by 1980 the overall structure of the receptor had been determined by protein chemistry to comprise a pentamer containing four subunits designated α2βγ∂ (e.g. Raftery et al, 1980).
In 1982, using DNA probes derived from a partial amino acid sequence of the Torpedo receptor, Shosaku Numa and his colleagues cloned the full-length cDNA and deduced the complete amino acid sequence for the α-subunit of the Torpedo receptor (Noda et al., 1982); and in subsequent papers reported cDNAs and sequences for the other three subunits (Noda et al., 1983: Nature, 301, 251–255, and 302, 538–542).
Knowledge of the muscle receptor composition allowed the neural nicotinic receptors cDNAs to be isolated by homology screening from neural tissue (e.g. Boulter et al., 1986, see Dani, 2015; McGehee and Role, 1995 for others). Like muscle channels, neuronal channels are pentamers, but composed only of α and β subunits, or sometimes homomeric α-subunits. In mammalian neurons eight α-subunits (α1-α7, α9, and α10) and three β-subunits (β2-β4) have been identified. In the mammalian CNS, the most common combinations are α42β23, with two ACh binding sites at the α-β interfaces, or α43 β22, with potentially three binding sites, or five homomeric α7 subunits with up to five binding sites (Dani, 2015). Uniquely among neural receptors, the latter are blocked by bungarotoxins. They also have a fivefold higher calcium permeability than the α-β heteromers (and 10 times more than muscle receptors): this probably contributes to the presynaptic action of ACh (and nicotine) in enhancing transmitter release (McGehee and Role, 1996). The most prominent receptor in the peripheral nervous system (including sympathetic ganglia) is an α3β4 heteromer, though this is also present in the medial habenular and interpeduncular nuclei.
Knowing the genes allows the construction of knock-in or knock-out subunits. Cordero-Erausquin et al. (2000) summarise some of the effects of individual neuronal receptor subunit knock-outs in mice. As expected, sympathetic and autonomic functions are disrupted in α3 or β4 null mice. Deletion of α4 or β2 reduced high-affinity nicotine binding and some presynaptic transmitter release enhancing nicotinic receptors were non-functional in the β2 knockout mice. Furthermore, β2 subunits appeared to have a role in learning and in protection against ageing.
Looking at nicotinic receptors
An important early post-1967 advance was the discovery by C.Y. Lee of the snake venom toxin α-bungarotoxin, which binds irreversibly to muscle nicotinic receptors (Miledi and Potter, 1971). This not only facilitated the isolation and identification of the receptor but its tight binding allowed its use as a probe for localising the receptor at a much higher resolution than that that obtained with radiolabelled curare (Fertuck and Salpeter, 1974). The persistent binding of bungarotoxin also permitted experiments on end-plate receptor turnover and regulation (e.g. Levitt et al., 1980: Science, 210, 550–551).
The detailed atomic structure of the nicotinic ACh receptor has not yet been determined by X-ray crystallography, but the discovery of a secreted water-soluble ACh binding protein (Smit et al., 2001: Nature, 411, 261–268 has allowed the crystal structure of the homologous binding domain in the nicotinic receptor to be determined (Brejc et al., 2001).
On the other hand, the exceptional density and organisation of the receptors in Torpedo electroplax has been brilliantly exploited to provide images of the intact receptor down to 4Å resolution by cryo-electron microscopy (Unwin, 2013) – now much-favoured molecular imaging technique (see Fernandez-Leiro and Scheres, 2016: Nature, 537, 339–346).
What do neural nicotinic receptors do?
Other than the pre-1967 Renshaw cells, only a few functional cholinergic synapses with postsynaptic nicotinic receptors have yet been identified in the mammalian CNS (Jones et al., 1999). Instead, the majority of nicotinic receptors are presynaptic, and serve to enhance the release of other neurotransmitters such as glutamate (McGehee and Role, 1996) – very much as predicated from the experiments of Koelle and Nishi on sympathetic ganglia (see Box 1) (In some cases, this seemed truly surprising. Thus, as pointed out to us by Dr M.J.Brownstein (National Institutes of Mental Health (NIMH) one of the most striking “cholinergic” tracts in the brain (neurochemically speaking is the fasciculus retroflexus of Meynert (FRM), between the medial habenula and the interpeduncular nucleus (IPN). When we saw this gleaming white tract in dissected brain slices just begging to be stimulated, we thought this must be the perfect central homologue of the sympathetic ganglion synapse. But not so: although ACh and nicotinic agonists readily excited IPN neurons, the IPN response to FRM stimulation was not at all diminished by nicotinic antagonists; instead it was inhibited by glutamate antagonists, suggesting that transmission was glutamatergic (Brown et al., 1983: J.Physiol., 341, 655–670)!!. The most prominent effect of ACh or nicotinic agonists on FRM stimulation was to reduce the amplitude of the action potential recorded from the FRM terminals within the IPN and slow its conduction (Brown et al., 1984: J.Physiol. 353, 101–109; we suggested that this was due to a depolarization of the unmyelinated fibre terminals, as seen on peripheral C-fibres. In some elegant experiments, McGehee et al (1995: Science, 260, 1692–1696) later confirmed this presynaptic action, and showed that it led to an entry of Ca2+ through the nicotinic channels, and consequent enhancement of the glutamatergic epsc. More recently, Pen et al (2010: Neuron, 69, 445–452) have found that selective stimulation of choline acetyltransferase-expressing fibres in the FRM release both glutamate and ACh: glutamate drives the individual fast epscs in the IPN neurons while ACh released by repetitive 20–50 Hz FRM stimulation induces a lower amplitude slow nicotinic epsc in the IPN neurons.). Most of these presynaptic receptors are α4β2, occasionally α3β2 or β4 (e.g. medial habenula and interpeduncular nucleus, see footnote), or α7 homomers. Some clues regarding their overall functional significance may be gleaned from the effects of subunit knock-outs noted above – for example, disruption of some forms of learning, or loss of responses to nicotine such as antinociception (Cordero-Erausquin et al., 2000). Notwithstanding, there are very few (if any) examples of true cholinergic axo-axonal synapses, so presumably these presynaptic receptors are activated by more remotely released ACh–the ‘soup’ theory of transmission (Sivilotti and Colqhuoun, 1995: Science, 269, 1681–1682).
Muscarinic receptors (mAChRs)
The nature of the receptor
As with the nicotinic receptor, the physical nature of the muscarinic receptor was unknown in 1967. The first muscarinic receptor was cloned from a pig brain cDNA library by Kubo et al. (1986). The predicted amino acid sequence showed a clear homology to the β-adrenergic receptor (Dixon et al., 1986: Nature, 321, 75–79) and to the visual pigment rhodopsin (Ovchinnikov, 1982: FEBS Lett, 148 179–191) and hence it joined the family of heptahelical (7 transmembrane domain=7TM) signalling proteins.
Previous and ongoing pharmacological studies (e.g. Hammer et al., 1980) indicated that there may be more than one subtype of muscarinic receptor. Eventually, five genetic subtypes designated M1 through M5, were cloned (Bonner et al., 1987: Science, 237, 527–532; Fukuda et al., 1987: Nature, 327, 623–625; summarised in Bonner, 1989). Interestingly, each receptor is encoded by a separate intronless gene. The original pig brain receptor corresponded to the pharmacological M1 subtype described by Hammer et al (1980) – the most abundant muscarinic receptor expressed in the brain.
The structures of the M2 (Haga et al., 2012: Nature, 482 547–551) and M3 (Kruse et al., 2012: Nature, 482 552–556) receptors in their resting state have now been determined by X-ray crystallography, so that it is now possible to envisage the ligand-binding and G protein-binding domains, and the possible conformational changes accompanying ligand and G protein binding, in some detail (Hulme, 2013).
How do the receptors work?
Unlike nicotinic receptors, muscarinic receptors are not ion channels. Instead, they are members of the G protein-coupled receptor (GPCR) superfamily, that is, when activated by ACh, their usual (I say ‘usual’ because there is accumulating evidence that GPCRs can sometimes alternatively route through other associated proteins such as β-arrestins (DeFea, 2008: Br J Pharmacol, 153, 5298–5309). Primary response is to dock onto, and activate, a trimeric guanine nucleotide-binding protein called a G protein (Oldham and Hamm, 2008). G proteins were discovered in the 1970s through a requirement for guanosine triphosphate (GTP) in the solution when studying GPCR activity in broken cell preparations (Rodbell et al., 1971: J.Biol.Chem., 246, 1877–1882).
The G protein comprises α, β, and γ subunits. The α-subunit contains the guanine nucleotide binding domain, and also a GTPase catalytic domain. At rest the α-subunit binds guanosine diphosphate (GDP). The activated GPCR induces a conformational change in the G-protein leading to (a) dissociation of the trimer into α- and coupled βγ-subunits and (b) dissociation of GDP and its replacement by GTP. Hydrolysis of GTP by the GTPase activity of the α-subunit leads to replacement of GTP by GDP and reassembly of the αβγ trimer. The GTPase activity determines the rate of recovery, and may be accelerated by ancillary GTPase activating proteins (GAPs). The GPCR-induced reduction in the binding affinity of GDP to the α-subunit is matched by a reciprocal reduction in the apparent binding affinity of the agonist for the GPCR (shown for muscarinic receptors by Berrie et al. (1979: Biochem. Biophys. Res. Comm., 87 1000–1005).
There are a number of different G proteins, differentiated in terms of the structures and downstream targets of their α-subunit. The individual muscarinic receptors show a general pattern of G protein ‘preferences’ as follows (Bonner, 1989; Caulfield, 1993):
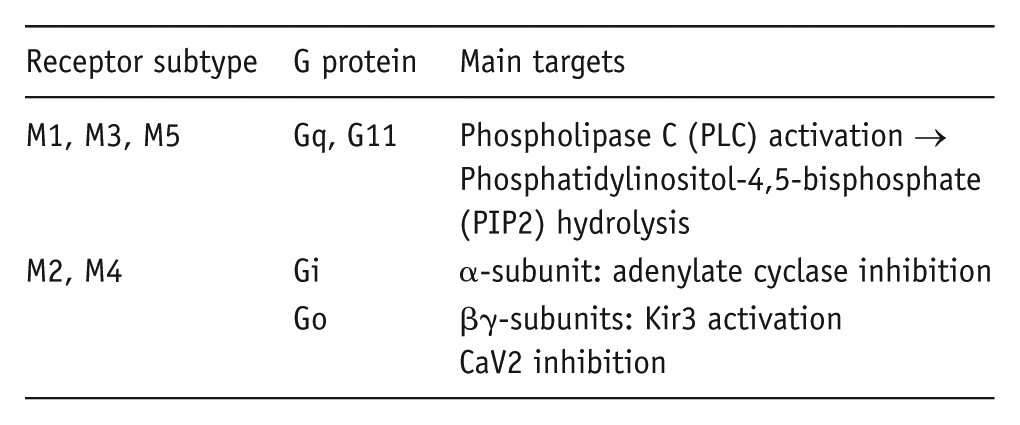
What do they do to a neuron?
In the short term, activation of mAChRs modifies the signalling properties of neurons by altering the activity of selected membrane ion channels using the apposite G protein (or one of its downstream biochemical effectors) as the receptor – ion channel transducer (see Brown, 2010; Caulfield, 1993 for reviews.) Thus, in brief, and with considerable simplification, activation of M1/M3/M5 receptors tends to increases neuronal excitability by inhibiting one or more of several potassium channels and/or by activating cation channels; whereas activation of M2 or M4 receptors produces postsynaptic inhibtion by activating Kir potassium channels, or presynaptic inhibition by inhibiting CaV2 calcium channels. However, the indirect nature of the pathway connecting receptor to ion channel mitigates against any hard and fast rules linking the receptor to the response, for the following reasons.
The ion channel only ‘sees’ the final transducer, not the receptor, so in principle cannot tell whether (say) Go has been activated by a muscarinic receptor or a metabotropic glutamate receptor; or, if the former, by the M2 or M4 receptor. Dissecting or predicting the final response and its mechanistic pathway when (say) ACh is applied to a neuron or a mixture of ACh and glutamate are released onto a neuron then becomes a question of anatomy: that is, which receptor/G protein/intermediary transducer/ion channel is/are present in that neuron, and whereabouts are they located in the neuron. For the latter, one might think first of which compartment of the neuron houses the receptors and channels. Thus, the CaV2 calcium channels closed by stimulating M2 or M4 receptors (and thence by Goβγ-subunits) are heavily concentrated in the presynaptic terminals of central and peripheral neurons, where they drive action potential-evoked transmitter release; so the principal effect of M2 or M4 receptor stimulation is to inhibit transmitter release – either of ACh itself (feedback auto-inhibition, for example, from basal forebrain axons: Allen and Brown, 1996: J.Physiol., 492 453–466), or of other transmitters (hetero-inhibition). On the other hand, the Kir3 channels opened by M2 or M4 receptors are primarily postsynaptic and generate a form of postsynaptic inhibition. As an example of subcellular compartmentation: in hippocampal neurons the Kv7 channels inhibited by M1 receptor activation are localised to the axon initial segment where they bind to ankyrinG and control the action potential threshold; hence, their inhibition by ACh increases excitability by facilitating local spike generation (Martinello et al., 2015; Shah et al., 2008: Proc.Natl.Acad.Sci.,USA, 105, 7869–7874). In other neurons where the Kv7 channels are somatic, their inhibition by mAChR receptors can also increase excitability more generally, by depolarising the cell and increasing input resistance. Further micro-anatomical association and segregation of muscarinic receptors with their cognate G proteins and ion channels may be achieved by association with ancillary scaffolding proteins such as A-kinase Anchoring Proteins (AKAPs; Kosenko et al., 2012: EMBO J., 31, 3147–3156).
Unlike nicotinic receptors, or other transmitter-gated ionotropic receptors, the response to stimulating muscarinic receptors is indirect and takes time, from about 30–50 ms for the activation of a G protein-gated inward rectifier Kir3 potassium channel by an M2 receptor, to ⩾200 ms for the closure of an M-type Kv7 potassium channel by an M1 receptor (via Gq and consequent fall in membrane PIP2 concentration: Hille et al., 2014), seconds or minutes for responses involving downstream phosphorylation or dephosphorylation, and several hours for transcription changes (e.g. a change in the number of M-type channels induced by a calcium-dependent transcriptional response to neural excitation by M1-mAChRs (Zhang and Shapiro, 2012: Neuron. 76 1133–1146).
Muscarinic receptors and global nervous system function
Thiele (2013) provides a contemporary update on the contributions of muscarinic receptors to central nervous system physiology. Wess (2004) summarises the roles of the individual muscarinic receptors to nervous system function in mice as revealed by genetic subtype deletions. A few points of interest from these surveys:
As predicated by Krnjevic (1967, 1974 (see Box 1) a major contribution of muscarinic excitation of cortical and hippocampal neurons to cortical arousal and cognition has been established. This is mediated substantially by M1 receptors, but probably with additional input from M5 receptors, and with a negative feedback effect from presynaptic M2 autoreceptors onto ascending cholinergic afferents.
Substantially more information about the cellular mechanisms of muscarinic excitation of cortical and hippocampal neurons and their consequences for network behaviour has been obtained over the past 50 years (see also Brown, 2010; Martinello et al., 2015: 346–353).
Also, as predicated from old pharmacological knowledge, activation of muscarinic receptors affects basal ganglion locomotor function, probably through release of ACh from striatal interneurons and indirect inhibition of dopamine release by M4 receptors. An additional role for M1 receptors is indicated by an increase in striatal dopamine levels in M 1R k-o mice.
A cholinergic input also enhances dopamine release in the ventral tegmental ‘reward centre’ through an action on M5 receptors. This is subject to M4 auto-inhibition of ACh release, so dopamine levels in the nucleus accumbens are raised in M4 k-o mice.
Muscarinic agonists induce an analgesic effect of supraspinal origin when injected intra-thecally. This is mediated by a combination of M2 and M4 receptors, offering interesting prospects for drug development.
Future prospects
With increasingly precise information about the structure of the receptors, we might expect the development of increasingly selective drugs targetting different subtypes of muscarinic receptors and subtype combinations of nicotinic receptors.
For the muscarinic receptors, subtype selectivity is likely to be best achieved by targetting allosteric sites (see Conn et al., 2009: Trends Pharmacol Sci., 30: 148–155). Apart from greater selectivity, positive alllosteric modulators (PAMS) have the advantage over direct agonists that they only affect ongoing cholinergic activity. Thus, M1-PAMS show promise for treatment of cognitive disorders while M4-PAMS may be appropriate for treatment of schizophrenia.
In the past, much work on nicotinic receptor pharmacology has been driven by the need to control nicotine addiction, but this will be a declining market in the future. Nevertheless, drugs interacting with nicotinic receptors may have beneficial effects independently of nicotine actions. For example, PAMs for α7 nicotinic receptors have beneficial effects on cognition and pain in animal studies (Bagdas et al., 2017: Br. J. Pharmacol., 173(16): 2506–2520; Potasiewicz et al., 2017: Neuropharmacol, 113: 188–197).
Optogenetic techniques coupled with refined recording of neuronal responses in vivo should allow a much more precise description of the cholinergic circuits underlying the behavioural responses to cholinergic stimulation in the brain and of their effect on neural coding, to the extent that they might be simulated in virtual reality and the effects of pathological lesions in (e.g.) Alzheimer’s Disease and their pharmacological amelioration fully understood.
Footnotes
Acknowledgements
I thank Dr Susan Jones (University of Cambridge) for helpful comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.