
Abstract
Rationale:
Adjunct intra-arterial alteplase has been shown to potentially improve clinical outcomes in patients with large vessel occlusion (LVO) stroke who have undergone successful endovascular thrombectomy. Tenecteplase, known for its enhanced fibrin specificity and extended activity duration, could potentially enhance outcomes in stroke patients after successful reperfusion when used as an adjunct intra-arterial therapy.
Aim:
To explore the safety and efficacy of intra-arterial tenecteplase after successful endovascular thrombectomy in patients with LVO stroke.
Sample size:
To randomize 498 participants 1:1 to receive intra-arterial tenecteplase or no intra-arterial adjunctive thrombolysis therapy.
Methods and design:
An investigator-initiated, prospective, randomized, open-label, blind-endpoint multicenter clinical trial. Eligible patients with anterior circulation LVO stroke presenting within 24 h from symptom onset (time last known well) and excellent to complete reperfusion (expanded Thrombolysis In Cerebral Infarction (eTICI) scale 2c–3) at endovascular thrombectomy are planned to be randomized.
Outcomes:
The primary outcome is freedom from disability (modified Rankin Scale, mRS, of 0–1) at 90 days. The primary safety outcomes are mortality through 90 days and symptomatic intracranial hemorrhage within 48 h.
Discussion:
The POST-TNK trial will evaluate the efficacy and safety of intra-arterial tenecteplase in patients with LVO stroke and excellent to complete reperfusion.
Introduction and rationale
Endovascular thrombectomy (EVT) has been established as the standard treatment for patients with acute large vessel occlusion (LVO) stroke.1–6 However, despite advancements in thrombectomy devices and workflow processes, the outcomes for patients with LVO continue to remain less than optimal. Notably, while excellent to complete reperfusion (expanded Thrombolysis In Cerebral Infarction (eTICI) scale 2c–3) can be achieved in more than 50% of patients with LVO, only half of patients achieve freedom from disability (modified Rankin Scale [mRS] 0–1) at 90 days.7,8 This incomplete recovery may be partly due to infarction that occurred prior to the intervention but also partly due to progression of infarct after intervention into regions with insufficient macrocirculatory and microcirculatory reperfusion.9,10 Previous studies have demonstrated that hypoperfusion was common (ranging from 25.3% to 58%) in patients treated with EVT despite successful reperfusion, which was associated with expanding infarct and lower odds of functional independence. 11 Among patients with an eTICI 2c technical outcome, 1%–10% of the target artery territory remains hypoperfused due to visible distal emboli. In addition, in patients with eTICI 3 technical outcome (no visible occlusions present), hypoperfusion can occur due to obstruction in the microcirculation.
Adjunctive thrombolytic agents can dissolve macrothrombi and microthrombi to prevent ischemic damage by improving reperfusion. In routine clinical practice, intra-arterial thrombolysis is typically employed as a rescue therapy following unsuccessful or incomplete EVT, or for the treatment of thrombi arising during the procedure in newly affected.12–14 However, recently the CHOICE (Chemical OptImization of Cerebral Embolectomy) phase IIb randomized trial showed that adjunct intra-arterial alteplase could potentially improve the proportion of freedom from disability at 90 days without an increased risk of symptomatic intracranial hemorrhage (sICH) in patients with LVO stroke and successful reperfusion following EVT. 15 In addition, a brain imaging substudy of the CHOICE trial found that adjunct intra-arterial alteplase could enhance reperfusion and reduce the expansion of infarction, which may lead to better functional outcome. 11
Tenecteplase, compared with alteplase, is characterized by greater fibrin specificity, a longer half-life, and ease of administration as a single bolus. Some previous studies have showed that intravenous tenecteplase is associated with better reperfusion and functional outcome than alteplase in patients with acute ischemic stroke (AIS). 16 A secondary analysis of the EXTEND-IA TNK trial showed that tenecteplase had higher early reperfusion compared to alteplase in patients who had low clot burden. 17 Accordingly, studies with adequate sample size are warranted to inform whether adjuvant intra-arterial tenecteplase in patients with successful reperfusion may confer improved patient outcomes.
We initiated the Adjunctive Intra-arterial Tenecteplase after Successful Endovascular Thrombectomy in Patients with Large Vessel Occlusion Stroke (POST-TNK) trial to explore the efficacy and safety of adjuvant intra-arterial tenecteplase in patients with LVO stroke following excellent to complete reperfusion after EVT.
Methods
Study design
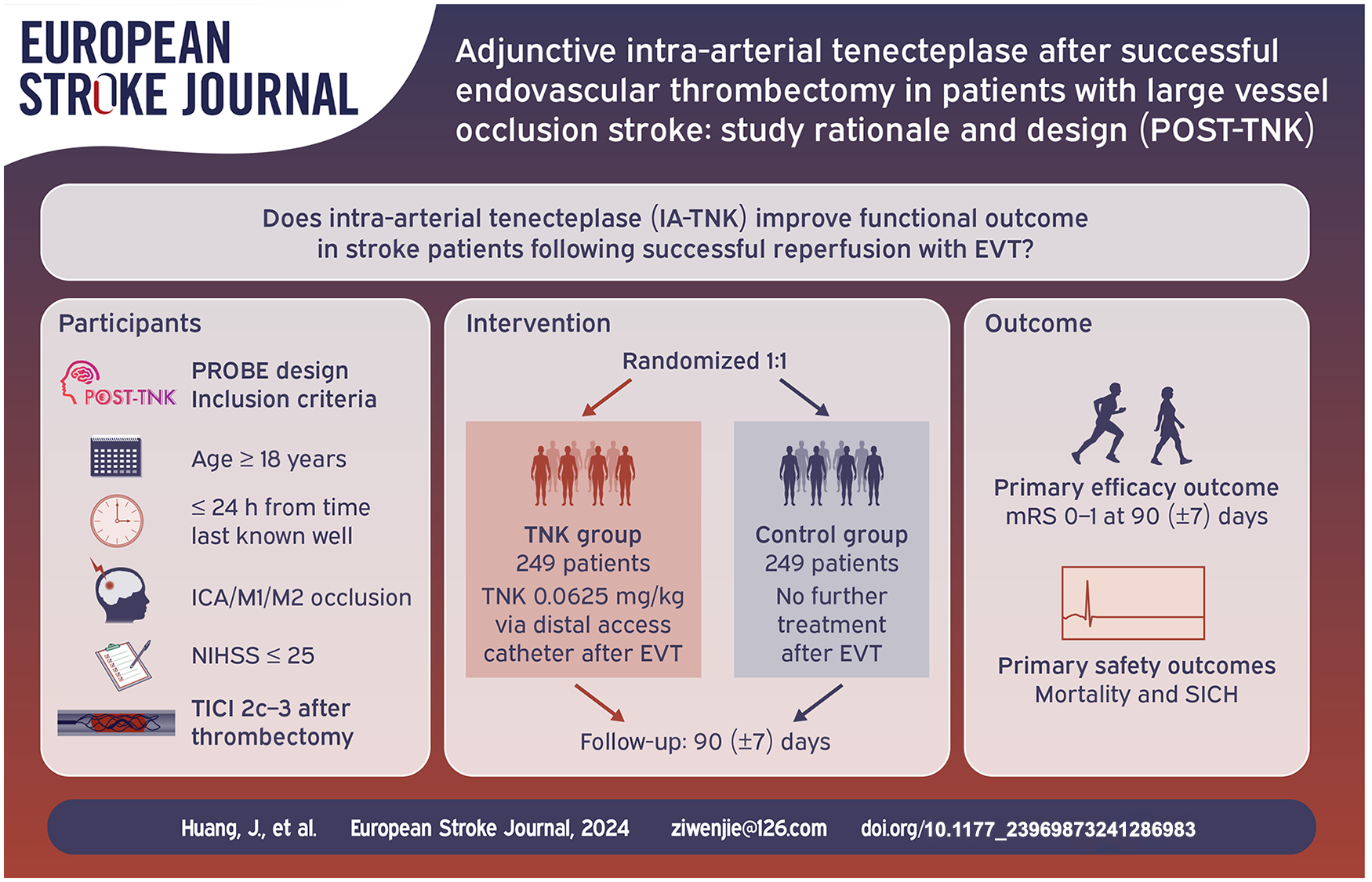
The POST-TNK trial is a multicenter, prospective, randomized, open-labeled, blinded outcome clinical trial (Figure 1). This trial was registered at www.chictr.org.cn (ChiCTR2200064809). The trial was designed in compliance with the Declaration of Helsinki. The protocol was approved by the ethics committee of the Second Affiliated Hospital of Chongqing Medical University and all participating centers. This study included 34 stroke centers in China. A qualifying participating center must have met the requirement of performing a minimum of 60 EVT procedures annually using stent-retriever or contact aspiration devices. Additionally, all neuro-interventionalists should possess a minimum of 3 years of experience in neuro-intervention and have completed at least 50 EVT procedures. All endpoints of this study will be evaluated by independent investigators who are unaware of the treatment allocation and the actual treatment received. The trial flowchart is in Figure 2.

Trial logo.

Study flowchart.
Participant population
Inclusion criteria include:
Aged ⩾ 18 years;
Presenting with AIS symptoms within 24 h from symptom onset (time last known well);
Occlusion of the intracranial internal carotid artery, M1 segment or M2 segment of middle cerebral artery (MCA);
Baseline National Institutes of Health Stroke Scale (NIHSS) ⩽ 25;
Baseline Alberta Stroke Program Early CT Score (ASPECTS) ⩾ 6 based on non-contrast computed tomography (NCCT) if the onset time within 6 h; ASPECTS ⩾ 7 or meet the Endovascular Therapy Following Imaging Evaluation for Ischemic Stroke (DEFUSE 3) study criteria (infarct volume of less than 70 ml, ratio of volume of 1.8 or more, and an absolute penumbra volume of 15 ml or more) or meets the DWI or CTP Assessment with Clinical Mismatch in the Triage of Wake-Up and Late Presenting Strokes Undergoing Neurointervention with Trevo (DAWN) study criteria (I: aged ⩾ 80 years, NIHSS ⩾ 10, infarct volume < 21 ml; II: aged < 80 years, NIHSS ⩾ 10, infarct volume < 31 ml; III: aged < 80 years, NIHSS ⩾ 20, infarct volume of 31 to less than 51 ml) within 24 h;
Treated with EVT resulting in excellent to complete reperfusion (eTICI 2c–3);
Informed consent obtained from patients or their legal representatives.
Exclusion criteria include:
Intracranial hemorrhage confirmed by computed tomography (CT) or magnetic resonance imaging (MRI);
Treated with intravenous thrombolysis or contraindication to intravenous thrombolysis (except time to therapy);
mRS score ⩾ 2 before stroke onset;
Vascular rupture, dissection, or contrast extravasation during the procedure;
Procedure time > 90 min;
Number of thrombectomy devices > 3;
Pregnant or lactating women;
Allergic to contrast agents;
Participating in other clinical trials;
Systolic blood pressure > 185 mmHg or diastolic blood pressure > 110 mmHg, refractory to antihypertensive drugs;
Known genetic or acquired bleeding diathesis, lack of anticoagulant factors, or oral anticoagulants with INR > 1.7;
Blood glucose < 2.8 mmol/l (50 mg/dl) or >22.2 mmol/l (400 mg/dl), platelets < 90 × 109/l
Bleeding history (gastrointestinal or urinary tract bleeding) in prior 1 month;
Chronic hemodialysis and severe renal insufficiency (glomerular filtration rate < 30 ml/min or serum creatinine > 220 μmol/L [2.5 mg/dl]);
Life expectancy due to any advanced disease < 6 months;
Follow-up is not expected to be completed;
Intracranial aneurysm or arteriovenous malformation;
Brain tumors with imaging mass effect;
Complete clinical recovery after rapid reperfusion.
Randomization
After confirming patient eligibility and receiving informed consent, randomization will be conducted through a web-based application (www.jinlingshu.com). Eligible patients will be randomly assigned to either the intra-arterial tenecteplase or control group in a 1:1 ratio.
Treatments
Both treatment groups will undergo EVT, which includes techniques such as stent retrievers, aspiration, balloon angioplasty, stenting, or any combination. Eligible patients assigned to the tenecteplase group will undergo an infusion of intra-arterial tenecteplase 0.0625 mg/kg, with a maximum dose of 6.25 mg, and administered over 10–15 min. This infusion will be administered through a distal access catheter or microcatheter positioned proximal to the initially occluded artery. Patients allocated to the control group will terminate the procedure without further intra-arterial thrombolysis.
Efficacy endpoints
The primary endpoint is freedom from disability defined as modified Rankin Scale (mRS) score of 0–1 at 90 (±7) days after randomization.
The secondary endpoints are:
(1) mRS score 0–2 at 90 (±7) days;
(2) level of disability (shift analysis of mRS score) at 90 (±7) days;
(3) The change of the NIHSS score at 5–7 days or discharge if earlier from baseline;
(4) European Quality Five-Dimension scale score at 90 (±7) days.
Safety endpoints
The primary safety endpoints are mortality at 90 (±7) days, symptomatic intracranial hemorrhage (sICH) within 48 h according to the modified Heidelberg Bleeding Classification.18,19 Other safety outcome includes any intracranial hemorrhage within 48 h.
Data and Safety Monitoring Board
An independent Data and Safety Monitoring Board (DSMB) will be established, comprising three experts – a neurologist, a neurointerventionalist, and a biostatistician independent of the overall study statistician. The DSMB members will not be participants in the trial or have any affiliation with the study sponsors. The board will convene annually to oversee the progress of the trial. Furthermore, the DSMB will review the occurrence of serious adverse events to guarantee the safety of the patients.
Sample size estimate
We assumed freedom from disability (mRS 0–1) rates of 32.8% in the control group based on the proportion of patients who achieved eTICI 2c–3 in the Endovascular Treatment With versus Without Tirofiban for Stroke Patients With Large Vessel Occlusion (RESCUE BT) trial. 20 As an 18.6% absolute difference of the mRS 0–1 was reported in the CHOICE trial, we conservatively estimated a 13% difference between the two groups, which meant that the proportion of mRS 0–1 in the tenecteplase group would be 45.8%. A sample size of 472 (236 per arm) patients would provide 83% power at a two-sided significance level of 0.05. Considering a 5% attrition rate, a total of 498 patients would be required (249 per arm).
Statistical analysis
The primary endpoint will be analyzed with a generalized linear model (GLM), from which risk ratio and its 95% confidence interval will be derived. The treatment effect will be estimated adjusted for the following prognostic variables including age, baseline NIHSS, baseline ASPECTS, time from onset to randomization, and occlusion location. The secondary outcomes and safety outcomes will be analyzed similarly using GLMs or assumption-free method. 21 Primary data analyses will be based on the intention-to-treat principle. The per-protocol analyses will also be performed as supplemental analyses. All statistical analyses will be performed using SAS version 9.4 (SAS Institute) and R version 4.3.0 (R Foundation for Statistical Computing). The trial results will be reported following the Consolidated Standards of Reporting Trials guidelines for reporting randomized trials. All analyses will be detailed in the statistical analysis plan which will be finalized before the unblinding of the study data.
Study organization and funding
The POST-TNK is an investigator-initiated clinical trial. It is sponsored by the Second Affiliated Hospital of Chongqing Medical University and the trial is supported by grants from the National Natural Science Foundation of China (No. 82425021, 82271349, 82071323), Chongqing Technology Innovation and Application Development Project (No. CSTB2022TIAD-KPX0160). CSPC Pharmaceutical Group Limited offered the study drugs only. The funders had no role in the design, planning or conduct of the trial and they will have no role in the analysis of the trial data, the writing of the manuscript or the interpretation of the trial data.
Trial status
This study was registered on October 19, 2022, and recruitment began on October 26, 2022. The enrollment was finished on March 2024.
Discussion
In the era of EVT for LVO stroke, excellent to complete reperfusion is now achieved in more than half of patients. In these patients, the critical focus is on improving patient outcomes after EVT. 22 Macrovascular reperfusion without microvascular reperfusion is known as the no-reflow phenomenon, which is estimated to occur in approximately a third of patients.23,24 To our knowledge, the POST-TNK trial, with a planned enrollment of 498 participants, is currently the largest registered study to explore the efficacy and safety of adjuvant intra-arterial tenecteplase in patients with LVO stroke after excellent to complete reperfusion.
The POST-TNK trial enrolled the first patient on October 26, 2022 and finished patient enrollment on March 2024. Data collection and queries are ongoing. When completed, this trial will provide pivotal data for evaluating the efficacy and safety of adjunctive intra-arterial tenecteplase in patients with anterior circulation LVO stroke and successful recanalization. Additionally, at least four similar studies are under way that will provide more evidence of the feasibility and the safety of intra-arterial thrombolysis following successful recanalization in patients with LVO stroke: Intra-arterial Alteplase for Acute Ischemic Stroke After Mechanical Thrombectomy (PEARL, NCT05856851); CHemical OptImization of Cerebral Embolectomy 2 (CHOICE 2, NCT05797792); Intra-arterial Recombinant Human TNK Tissue-type Plasminogen Activator Thrombolysis for Acute Large Vascular Occlusion After Successful Mechanical Thrombectomy Recanalization (ANGEL-TNK, NCT05624190) and Post-thrombectomy Intra-arterial Tenecteplase for Acute manaGement of Non-retrievable Thrombus and No-reflow in Emergent Stroke (EXTEND-AGNES TNK, NCT05892510).
In the POST-TNK trial, the dose of TNK administered intra-arterially was one quarter of the intravenous TNK thrombolytic dose in stroke, a method comparable to the CHOICE trial. In addition, the ATTENTION IA trial evaluated the use of intra-arterial tenecteplase 0.0625 mg/kg after successful EVT of an arterial occlusion of the vertebral, basilar or P1 segment of the posterior cerebral artery.25,26 We did not include patients who had received intravenous thrombolysis, to mitigate hemorrhage risk with additional intra-arterial thrombolytic.
We acknowledge limitations to our clinical trial. Our sample size estimate was based on the phase IIb CHOICE randomized trial that was terminated early for administrative reasons. As the effect size for the primary outcome of mRS 0–1 was large in the CHOICE trial (18.6%), it is possible that even our conservative estimate of a 13% treatment effect may be too wide, and our sample size may be underpowered.
Conclusions
The POST-TNK trial will evaluate the efficacy and safety of intra-arterial tenecteplase in patients with LVO stroke and excellent to complete reperfusion.
Supplemental Material
sj-docx-1-eso-10.1177_23969873241286983 – Supplemental material for Adjunctive intra-arterial tenecteplase after successful endovascular thrombectomy in patients with large vessel occlusion stroke (POST-TNK): Study rationale and design
Supplemental material, sj-docx-1-eso-10.1177_23969873241286983 for Adjunctive intra-arterial tenecteplase after successful endovascular thrombectomy in patients with large vessel occlusion stroke (POST-TNK): Study rationale and design by Jiacheng Huang, Changwei Guo, Jie Yang, Xiaolei Shi, Chang Liu, Jiaxing Song, Fengli Li, Weilin Kong, Shitao Fan, Zhouzhou Peng, Shihai Yang, Jinfu Ma, Xu Xu, Linyu Li, Zhixi Wang, Nizhen Yu, Wenzhe Sun, Chengsong Yue, Xiang Liu, Dahong Yang, Cheng Huang, Duolao Wang, Raul G Nogueira, Thanh N Nguyen, Jeffrey L Saver, Yangmei Chen and Wenjie Zi in European Stroke Journal
Footnotes
Acknowledgements
None.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JLS reports consulting fees for advising on rigorous and safe clinical trial design and conduct from Biogen, Boehringer Ingelheim, Genentech, Johnson&Johnson, Phenox, Phillips, Rapid Medical, and Roche. TNN discloses Associate Editor of Stroke, advisory board of Aruna Bio and, Brainomix. RGN reports consulting fees for advisory roles with Anaconda, Biogen, Cerenovus, Genentech, Philips, Hybernia, Hyperfine, Imperative Care, Medtronic, Phenox, Philips, Prolong Pharmaceuticals, Stryker Neurovascular, Shanghai Wallaby, Synchron, and stock options for advisory roles with Astrocyte, Brainomix, Cerebrotech, Ceretrieve, Corindus Vascular Robotics, CrestecBio Inc., Euphrates Vascular, Inc., Vesalio, Viz-AI, RapidPulse and Perfuze. RGN is one of the Principal Investigators of ENDOLOW trial. Funding for this project is provided by Cerenovus. RGN is the Principal Investigator of the DUSK trial. Funding for this project is provided by Stryker Neurovascular. RGN is an investor in Viz-AI, Perfuze, Cerebrotech, Reist/Q’Apel Medical, Truvic, Tulavi Therapeutics, Vastrax, Piraeus Medical, Brain4Care, Quantanosis AI, and Viseon. Other author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author(s) disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This clinical trial is sponsored by (1) National Natural Science Foundation of China (No. 82425021, 82271349, 82071323), Chongqing Technology Innovation and Application Development Project (No. CSTB2022TIAD-KPX0160). (2) CSPC Pharmaceutical Group Limited, China (study drug only). The sponsors had no role in the study design, data collection, analysis, and interpretation, and in drafting and submitting this article.
Ethical approval
This study involves human participants and was approved by the ethics committee of the Second Affiliated Hospital of Chongqing Medical University and all participating centers.
Informed consent
Informed consent obtained from patients or their legal representative.
Guarantor
Wenjie Zi
Contributorship
JCH, CWG, DLW, RGN, TNN, JLS, YMC, and WJZ designed and conceptualized the study. JY, XLS, CL, JXS, FLL, WLK, STF, ZZP, SHY, JFM, XX, LYL, ZXW, NZY, WZS, CSY, XL, DHY, and CH participated in data collection. JCH and CWG wrote the manuscript. All authors critically revised and approved the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.