
Abstract
Rationale:
A large proportion of stroke survivors will have long-lasting, debilitating neurological impairments, yet few efficacious medical treatment options are available. Etanercept inhibits binding of tumor necrosis factor to its receptor and is used in the treatment of inflammatory conditions. Perispinal subcutaneous injection followed by a supine, head down position may bypass the blood brain barrier. In observational studies and one small randomized controlled trial the majority of patients showed improvement in multiple post stroke impairments.
Aim:
Perispinal Etanercept to improve STroke Outcomes (PESTO) investigates whether perispinal subcutaneous injection of etanercept improves quality of life and is safe in patients with chronic, disabling, effects of stroke.
Methods and design:
PESTO is a multicenter, international, randomized placebo-controlled trial. Adult participants with a history of stroke between 1 and 15 years before enrollment and a current modified Rankin scale between 2 and 5 who are otherwise eligible for etanercept are randomized 1:1 to single dose injection of etanercept or placebo.
Study outcomes:
The primary efficacy outcome is quality of life as measured using the Short Form 36 Health Inventory at day 28 after first injection. Safety outcomes include serious adverse events.
Sample size target:
A total of 168 participants assuming an improvement of the SF-36 in 11% of participants in the control arm and in 30% of participants in the intervention arm, 80% power and 5% alpha.
Discussion:
PESTO aims to provide level 1 evidence on the safety and efficacy of perispinal etanercept in patients with long-term disabling effects of stroke.

Keywords
Introduction and rationale
Despite major advances in acute stroke care, at least 1-in-3 stroke survivors have long-lasting, debilitating neurological impairments. Consequently, many stroke survivors are unable to live at home, return to work or perform their usual activities or hobbies. Improving life after stroke is a key priority as recognized in the Stroke Action Plan for Europe 2018–2030 and the American Heart Association’s Recommendations.1–3 The efficacy of current medical treatment options in reducing post-stroke impairments is however limited and new approaches are therefore needed.
Etanercept is a human tumor necrosis factor (TNF) receptor p75 Fc fusion protein produced by recombinant DNA technology. Etanercept competitively inhibits TNF binding to cell surface TNF-Receptors, preventing TNF-mediated cellular responses by rendering TNF biologically inactive. 4 Etanercept may also modulate biologic responses controlled by additional downstream molecules that are induced or regulated by TNF. Etanercept is commonly used in rheumatoid arthritis or psoriasis. TNF-alpha has multiple physiological functions within the CNS and restoration of normalized TNF levels within the CNS may have multiple beneficial effects on synaptic function, neurotransmission, glutamate levels and long-term potentiation. Activated microglia are the main source of TNF in the CNS, and there is evidence that elevated TNF-alpha levels maintain microglia in an activated state, creating a positive feedback loop. 5 This may explain its long-term persistence and the potential benefits of anti-TNF alpha treatments, even several years after stroke. 6 Supportive evidence of ongoing microglial activation has been reported up to 2 years after stroke using [11C](R)-PK11195 PET. 7
Due to its high molecular weight, etanercept does not cross the blood brain barrier when given systemically. Although controversial, it has been proposed that etanercept may enter the central nervous system via valveless vertebral veins after perispinal subcutaneous administration followed by head down positioning.8–10 According to proponents of this mechanism, head- down tilt positioning is used to facilitate delivery of etanercept into the choroid plexus and cerebrospinal fluid (CSF) after its absorption into the cerebrospinal venous system as it has been demonstrated in basic science models that head-down tilt can increase intracerebral venous pressure and facilitate passage of plasma proteins into the CSF. Supportive empirical evidence for perispinal delivery entering the cerebrospinal venous system is from a case report with SPECT tracer. 8 CSF presence of etanercept after perispinal injection has been shown in rats using PET with radiolabeled etanercept. 9
Recently, the effects of perispinal etanercept administration have caught the attention of the stroke survivor community in Australia, Europe and the United States of America. Case reports, observational studies and one small, randomized trial in poststroke pain have described impressive and immediate improvements in gait, memory, pain, swallowing and muscle strength after perispinal subcutaneous administration.11–13 For instance, observational studies have shown highly significant improvements in grip strength reported immediately and up to at least 3 weeks after a single administration of perispinal etanercept. 12 Improvements in the time to walk 20 m were also noted. Ninety percent of the respondents reported favorable changes in motor effects after administration. 12 These effects were maintained over several weeks. In this published series, the effects were seen after a single administration. Anecdotal reports and the pilot study in poststroke pain have suggested that additional improvements are seen with repeated administration. 13
Regulatory authorities have not given approval for the treatment of poststroke disability with etanercept. In 2016, the American Academy of Neurology published a practice advisory which concluded that: “Research is needed on the effects of etanercept on poststroke disability. To minimize the risk of bias, future studies of etanercept for the treatment for poststroke disability should randomly allocate patients in a masked fashion to active and placebo treatment arms and employ masked, standardized poststroke disability outcome measures.” 14 In Australia, consumer groups lobbied Government to support clinician trials of etanercept for people with stroke. In response, the Australian Government created an open call for an etanercept trial and provided trial funds. An independent panel subsequently awarded the grant to the PESTO group. The Perispinal Etanercept to Improve Stroke Outcomes (PESTO) trial, is a phase 2b, multicenter, international clinical trial that will determine whether perispinal etanercept significantly improves quality of life 28 days after injection in chronic stroke survivors. We will test the safety and efficacy of perispinal etanercept administration in adult stroke survivors.
Methods
PESTO is a phase 2b, multicenter, international (Australia and Aotearoa New Zealand), prospective, randomized, placebo controlled, double-blind, parallel group trial with concealed allocation, blinded measurement and intention-to-treat analysis.
Patient population − inclusion and exclusion criteria
Adult stroke survivors aged up to 70 years will be enrolled between 1 and 15 years after their index ischemic or hemorrhagic event. The upper age limit was set based on the funding body’s call for the trial to be performed in adults of working age. Participants will be pre-screened using the 36-Item Short Form Health Survey v2 and are required to score a total of 95 or less out of a total of 100 to be considered eligible for the trial, and their modified Rankin Scale should be between 3 and 5, that is, with dependency. Participants with a modified Rankin scale of 2 (stroke leading to slight disability with inability to carry out all previous tasks can also be included provided their SF-36 score is below 80. Participants will undergo a pathology test to screen for a history of hepatitis B and C and tuberculosis. A full list of inclusion and exclusion criteria is provided in Table 1. The trial flow is shown in Figure 1.
Study eligibility inclusion criteria.

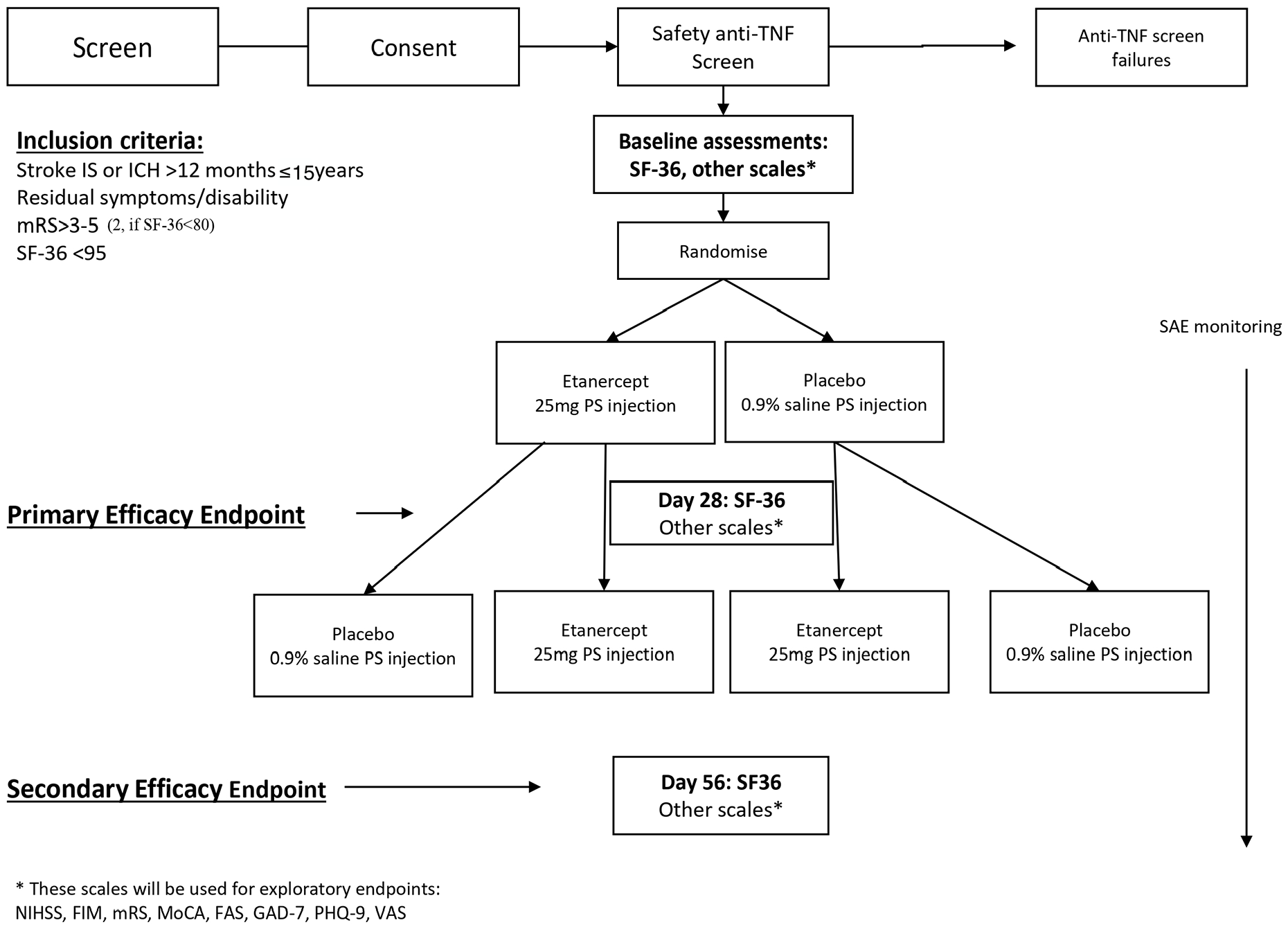
Flow of the trial.
Randomization
After confirmation of eligibility participants will first be randomized in 1:1 ratio to either receive a single dose of the etanercept (etanercept arm) or placebo (the control arm) using a centralized computer-generated random assignment procedure with permuted blocks of various sizes as part of the electronic case record form stored on a secure server based at the Florey. Randomization will be stratified by whether the pre-randomization SF-36 questionnaire was completed by the patient, or, if not possible, by a relative or carer proxy, and baseline modified Rankin Scale (dichotomized as mRS 2–3 vs mRS 4–5) and by time since stroke (1–5 years, 6–15 years after stroke onset). At the second step of the randomization procedure, the participants in the placebo arm will be subsequently randomized into receiving the second dose of placebo or a single dose of etanercept at day 28 after the first injection (based on 2:1 allocation ratio), while patients in the etanercept arm will be subsequently randomized into receiving a single dose of placebo or the second dose of etanercept at day 28 (based on 1:2 allocation ratio). This will result in three equally sized arms (Placebo n = 56, Single Dose etanercept n = 56, and Double Dose etanercept n = 56) for the secondary dose-response analysis of the efficacy outcome at day 56. (see Figure 2). A researcher not otherwise involved in the trial will undertake the randomization and allocation to treatment arms, thereby ensuring allocation concealment. The treatment allocation will be sent electronically to the study pharmacist who will prepare the investigational product. Study pharmacists will be instructed not to disclose treatment allocation. The investigator administering the investigational product, patients and outcome assessors will therefore be blinded to treatment allocation.
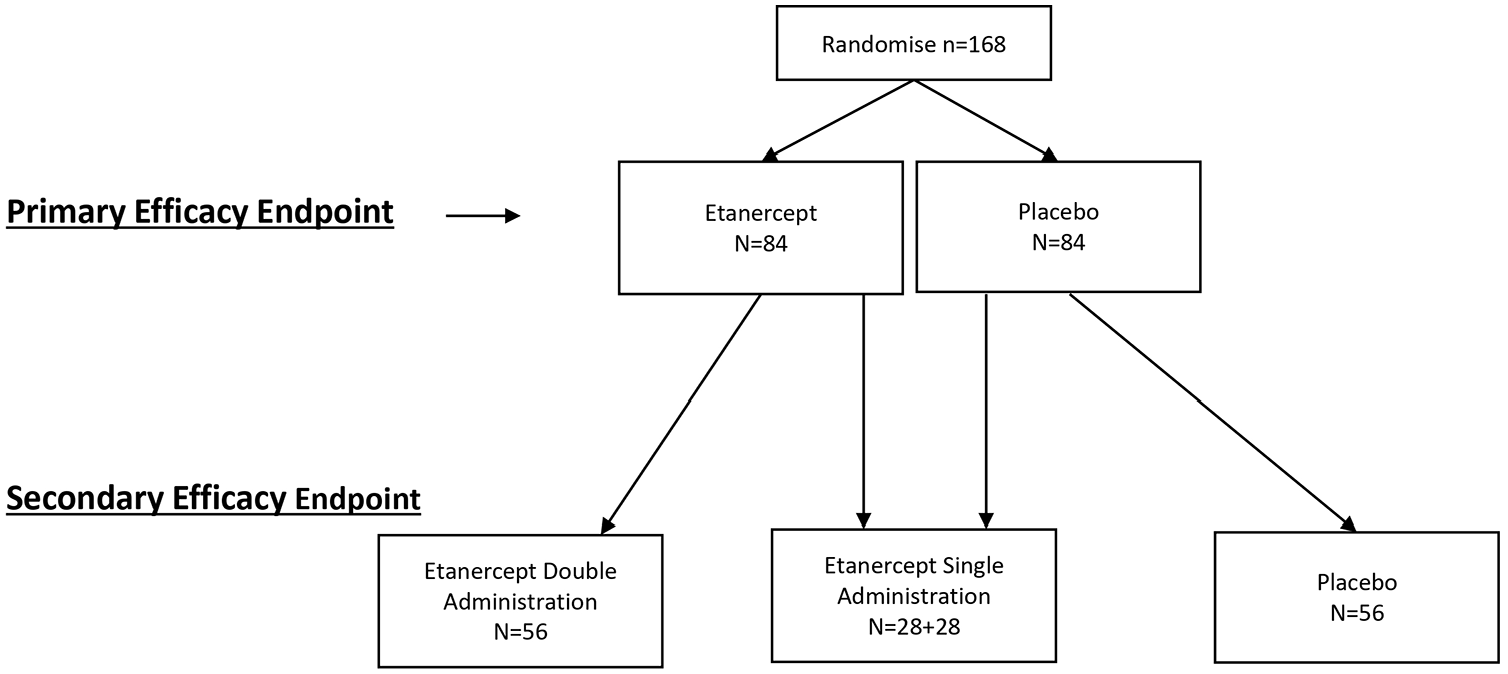
Dose-response analysis.
Treatment or intervention
Etanercept 25 mg lyophilized powder will be reconstituted with sterile water by an independent pharmacist at each participating site with sterile water for subcutaneous injection. The placebo control will be sterile saline (suitable for use as an injection) as a clear colorless solution and prepared in the same type of syringe with the same volume as etanercept. Given the possible slight discoloration of etanercept, syringes will be masked with a yellow tape. Active treatment or placebo, in an identical volume of 1.0 mL, will be administered via a posterior cervical interspinous (between the C6-C7 or C7-T1 interspace) injection subcutaneously in the midline with a 27-gauge half to one inch needle. The participant is then placed on an inversion table in the Trendelenburg position. The participant is laid supine, on a 20° incline with the feet elevated above the head for 4 min.
Primary outcomes
Primary efficacy endpoint
The proportion of participants at day 28 with an improvement of 5 points or more on the SF-36 compared to baseline following single injection of etanercept 25 mg or equivalent placebo. The change in SF-36 will be defined as the sum of changes in the Physical Component Summary score or the Mental Component Summary or both of the SF-36 between baseline and day 28.
Primary safety endpoints
Proportion of participants with serious adverse events at day 28.
Secondary outcomes
Secondary efficacy endpoint
The proportion of participants at day 56 with an improvement of 5 points or more on the SF-36 compared to baseline between the 3 treatment arms (placebo, single administration of etanercept, two administrations of etanercept).
Secondary safety endpoint
Exploratory endpoints
The National Institute of Health Stroke Scale (NIHSS), Functional Independent Measurement (FIM), modified Rankin Scale (mRS), Patient Global Improvement Scale (PGIC), Fatigue Assessment Scale (FAS), General Anxiety Disorder-7 (GAD-7), Patient Health Questionnaire (PHQ-9), Visual Analog Scale (VAS) for pain and Montreal Cognitive Assessment (MOCA) after injection of placebo or etanercept between baseline and day 28. Additionally, we will compare the NIHSS at day 56 in patients receiving none, one or two injections of etanercept.
Data monitoring body
An independent Data Safety Monitoring Committee (DSMC) including a statistician and two clinicians working in the area of stroke, will provide a blinded report to the management and Steering Committees according to a DSMC charter specifically outlining requirements.
Sample size estimates
Earlier work has shown that 11% of patients report a 5-point improvement in SF-36 when given placebo. 15 Based on the previous description of markedly significant improvement with etanercept in up to 90% of patients, for the primary efficacy outcome we anticipate that at day 28, 30% of the patients will have a 5-point improvement onSF-36. 12 Recruiting 160 patients (equally distributed between two arms) would yield 80% power to observe the absolute risk difference of 19%, assuming a two-tailed alpha of 0.05 and 11% of patients achieving a 5-point improvement in SF-36 when given placebo. To allow for potential dropouts, the proposed total sample size for this study is 168 patients, equally distributed between two arms.
Statistical analysis
A full statistical analysis plan will be completed prior to the study database lock. Analyses will be conducted on an intention-to-treat basis. The primary endpoint will be tested using a logistic regression model with achievement of at least 5 points improvement on the SF-36 compared to baseline as the dependent variable and with the treatment arm as an independent variable and baseline value of SF-36, proxy or self- administration of SF-36 as adjustment covariates.
Secondary and exploratory outcomes will be investigated using appropriate regression models with stratification variables and relevant baseline scores as adjustment covariates.
Study organization and funding
The study is funded by the Medical Research Future Fund (Australian Government Department of Health). The trial sponsor is the Florey Institute of Neuroscience and Mental Health. A management committee overseeing day to day operations is advised by a trial steering committee. Independent monitoring will be performed by a contract research organization.
Discussion
The current literature on perispinal etanercept is limited to uncontrolled, single center reports from the United States and one single center, phase I/II randomized trial in patients with chronic poststroke pain. This trial stopped early after 26 patients were enrolled and showed a reduction in pain levels measured by a visual analog scale. Trials evidence for this treatment is therefore limited.
PESTO is designed to include a representative sample of the patients that undergo off-label treatment with etanercept. In addition, the larger sample size, the multicenter nature and the focus on a quality-of-life measure will significantly expand the evidence base for this intervention. Nevertheless, the design of PESTO may be criticized and its conduct premature. We will include patients with severe disability with long standing stroke deficits. The pathophysiology of stroke strongly suggests that brain damage in these situations is irreversible. There is also a very limited understanding of the mechanism of action of TNF-alpha blockade in chronic stroke. The passage of etanercept using the perispinal route is also questionable. 10
Etanercept is reported to improve multiple post-stroke impairments (cognition, motor, sensory, and language), therefore impairment level outcomes scales such as the NIHSS or MOCA are less appropriate to assessing treatment efficacy. If we had chosen a specific impairment as an outcome measure for example, motor impairments we could have potentially missed meaningful improvements in other affected post stroke domains (mood, cognition, fatigue, and pain) Nevertheless, this information will be collected. The modified Rankin scale is traditionally used as an outcome parameter in acute stroke trials but given the narrow range of inclusion (mRS 2–5) and the low likelihood of observing changes in the mRS, we chose a patient reported outcome measure assessing quality of life as our primary outcome measure.16–18 The SF-36 is a robust measure of quality of life that has been used in multiple stroke trials and is very sensitive in detecting small improvements.19,20 Nevertheless, this choice may be criticized as SF-36 is self reported and more objective measurements at the level of impairment or functioning are used in most chronic stroke trials. We will report the NIHSS and mRS as additional outcome measures and will report change over time between treatment groups.
PESTO has other limitations. A health economic analysis plan is not included, and the trial will only be conducted in Australia and Aotearoa New Zealand Aotearoa and thus may not be broadly representative. Small treatment effects may be missed. Many questions remain on the potential mechanism of action of etanercept in alleviating chronic stroke impairments. The evidence supporting passage of etanercept into the central nervous system using the proposed mechanism is limited.
There are ethical and financial concerns regarding the conduct of trials based on insufficient experimental evidence. 21 We believe however that trials such as PESTO that answer questions raised by the stroke community, are the best way to inform the public, the stroke survivors’ community and regulators regarding efficacy and safety of perispinal etanercept. 22 A similar situation occurred with angioplasty of the jugular veins for the treatment of multiple sclerosis. After a long-standing debate, a randomized double-blind trial commissioned by health authorities provided conclusive evidence on the efficacy and safety of this intervention. 23
Conclusion
High quality efficacy and safety data based on randomized clinical trials are needed to inform optimal long term medical treatment after stroke. The results of PESTO will help determine whether further trials of perispinal etanercept are warranted.
Summary and conclusions
PESTO will determine whether perispinal subcutaneous etanercept is safe and improves quality of life in patients with long-term disabling effects of stroke.
Footnotes
Acknowledgements
The authors would like to acknowledge the Medical Research Future Fund (MRFF) for providing financial support for this study.
We are also grateful to the Florey institute of neurosciences, Austin Health, Alfred health, CGM research trust for providing resources and facilities for running the PESTO trial
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This is an academic investigator- initiated trial:
Vincent Thijs: VT reports no direct conflicts related to this publication. VT reports speaker fees for Amgen, Allergan, Astra Zeneca, Atricure, BMS, Bayer, Boehringer Ingelheim, Medtronic, unrelated to this publication. VT is on the editorial board for the following journals Annals of Neurology, Neurology, International Journal of Stroke and the European Stroke Journal. He is on the steering committee of the Librexia trial for which he receives modest compensation.
Geoffrey Cloud: None Nigel Gilchrist: None Brooke Parsons: None Forum Tilvawala: None Jan Ho: None
Lara Ruthnam: None Vimal Stanislaus: None Nikola Spriggs: None Marion Walker: None
Philip M Bath: PMB is Stroke Association Professor of Stroke Medicine and an emeritus NIHR Senior Investigator. He has consulted and received honoraria from CoMind, DiaMedica, Phagenesis and Roche.
Leonid Churilov: None Julie Bernhardt: None
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this research has been provided by the Medical Research Future Fund (MRFF). The MRFF has been established by the Australian Government to provide grants of financial assistance to support health and medical research and innovation, with the objective of improving the health and wellbeing of Australians. MRFF funding has been provided to Stroke Foundation under the MRFF Accelerated Research Program announced as part the MRFF’s disbursement package in October 2018. Further information on the MRFF and disbursements are available at ![]()
Ethical approval
Ethical approval for the study was granted by the Human Research Ethics committee(s) of the participating hospitals.
Informed consent
Written informed consent according to country-specific requirements is an inclusion criterion for the study.
Guarantor
Professor Vincent Thijs
Contributorship
All authors have contributed significantly to the study, meet the criteria for authorship, and have reviewed and approved the manuscript.
Trial registration
Name of trial registry: ACTRN12620001011976.