
Abstract
Purpose
Autophagy has emerged in recent years as a critical cellular survival mechanism for cell homeostasis and may play a protective role in atherosclerosis. We aimed to review here the role autophagy plays in different cell types present in carotid atherosclerotic plaques and that may be associated with the development of unstable carotid atheroma plaque.
Methods
We performed a thorough literature exploration in this area of research covering the three main cell types present in carotid atheroma plaques.
Findings
Reviewed reports indicate that the role of autophagy in stable or unstable carotid atherosclerotic plaques depends on the different cell types and phenotypes, the stage and morphology of the plaque and the specific autophagy factor/s involved.
Discussion
Although defective autophagy could be one of the causes for carotid atheroma plaques to become unstable, it is important to take into account that autophagic players can act differentially in different cell types and different stages of the developed plaque.
Conclusion
This review provides an overview of the role of autophagy in the main cell types in carotid atherosclerosis (i.e. macrophages, endothelial cells and smooth muscle cells).
Atherosclerosis in the carotid arteries is very common in the aging population. It is also one of the main risk factors to develop a cerebrovascular accident (CVA), 1 and recent research shows that CVA prevalence is increasing in the younger population. 2 CVA is one of the leading causes of morbidity/mortality and disability in the world with a high impact in societies. 3
Atherosclerosis is a chronic inflammatory disease that results in the progressive formation of atherosclerotic plaques. In this gradual process, several different cell types are implicated, and a sequence of events will take place that may lead ultimately to plaque rupture. The exact mechanisms involved in plaque rupture are not known but include lipid deposition, oxidative stress, inflammation, endothelial dysfunction, foam cell formation and smooth muscle cell (SMC) differentiation. In addition, plaque necrosis is a characteristic of the atherosclerotic lesions that cause cerebrovascular disease 4 and that are promoted by impaired phagocytic elimination of apoptotic cells.
Stenosis is the end result of atheroma plaque deposition in the artery. These atheroma plaques can be unstable, with a thin fibrous cap and a large fatty core (prone to rupture) or stable, with a thick fibrous cap and small fatty core (unlikely to rupture). Most clinicians correlate the instability of the atherosclerotic plaque with the development of cerebrovascular events. 5 Patients who suffer severe asymptomatic carotid stenosis higher than 70%–90% or symptomatic with stenosis higher than 70% would be routinely recommended for carotid endarterectomy (CEA) surgery. 6 Recently, due to the improved medical therapies, the benefit of CEA for asymptomatic patients has been questioned. 7 Although it is true that a proportion of patients with asymptomatic carotid disease will never become symptomatic, CEA would be crucial for those at high risk to become symptomatic. For that reason, understanding the molecular basis by which a plaque becomes unstable may be of relevance to identify patients who are at higher risk.
The mechanisms involved in plaque stability are complex, and the complete processes taking place are not completely understood. However, autophagy is known to play a role in the process of the atherosclerotic plaque development. 8 In this review, we will discuss current evidence for the role of autophagy in the development of an unstable atherosclerotic plaque.
Molecular basis of autophagy
Autophagy can be differentiated in three route types used to deliver cellular components to the lysosome: microautophagy, chaperone-mediated autophagy and macroautophagy. In this review, we focus mainly on macroautophagy, which routinely is termed as autophagy. It is a regulated mechanism required for cellular metabolism and homeostasis, and while basal autophagy is considered a housekeeping function to degrade intracellular harmful proteins and organelles, 9 excessive autophagy could lead to cell death. 10
Autophagy is an evolutionary conserved pathway regulated by autophagy-related genes (ATGs). It is a catabolic intracellular process associated with the formation of double-membrane structures called autophagosomes.
11
These are involved in the degradation of bulk cytoplasmic contents, aggregated or damaged proteins and damaged organelles. The material is transferred to the lysosomes by fusion with the autophagosomes, and degraded elements are subsequently released back into the cytosol where they can be recycled for biosynthesis or energy production. Autophagosome formation is driven by the cooperation of a group of proteins called autophagy-related proteins (abbreviated as ATGs) that can be classified into four groups (Figure 1). Autophagy initiation is controlled by the Atg1/Unc-51-like kinase (ULK) complex that is composed of ULK1/2, Atg13, FIP200 and Atg101 12,13 and can be regulated by different signals through the mTOR pathway (i.e. MAPK signalling, Akt signalling, ERK, TP53, PI3KI). Upon mTOR inhibition (i.e. activation of autophagy), mTOR is released from this complex and will subsequently activate the ULK kinase activity leading to the phosphorylation and activation of Atg13 and FIP200. Then, a complex composed of Beclin-1, class-III-phosphatidylinositol-3-kinase (PICK3C3) and Atg14, will be recruited by the ULK signalling pathway
14
leading to the activation of the first two ubiquitin-like pathways, which are responsible for vesicle formation.
15
The first of these, Atg12-Atg5 complex, is formed when Atg12 is activated by Atg7 and conjugates with Atg5 prior to the formation of a homodimer together with Atg16L1. This event is a requisite for the second ubiquitin-like pathway initiation (MAPLC3B (I/II)-phosphatidylethanolamine), which starts with the conjugation of MAP1LC3B-I to phosphatidylethanolamine through activation by Atg7, leading to the closure of the autophagosome formation. Once the autophagosome is completed, the Atg-5-Atg12-Atg16L complex dissociates and Atg4 releases MAP1LC3B-II thus allowing the fusion of the phagosome with the lysosome. Completion of this process requires the function of the lysosomal proteins LAMP2, LAMP1 and GTP-binding protein Rab7.
16
The degradation of the autophagic load, which will be released into the cytoplasm, takes place in these autophagolysosomes. Nevertheless, it is important to note that mTOR signalling constitutes not the only route towards activate autophagy, since for example inhibition of Inositol 1,4,5-Trisphosphate Receptor Type 1 (ITPR 1) provokes as well an up-regulation of autophagic activity by activating Activate Kinase Alpha 1 Sub-unit (AMPK) in a mTOR-independent manner,
17
which will activate the ULK complex. Other autophagy mTOR-independent routes can be revealed by autophagy regulators, which work thorough mTOR-independent pathways, such us threalose, spermidine, TLR7.18–20
Autophagy overview.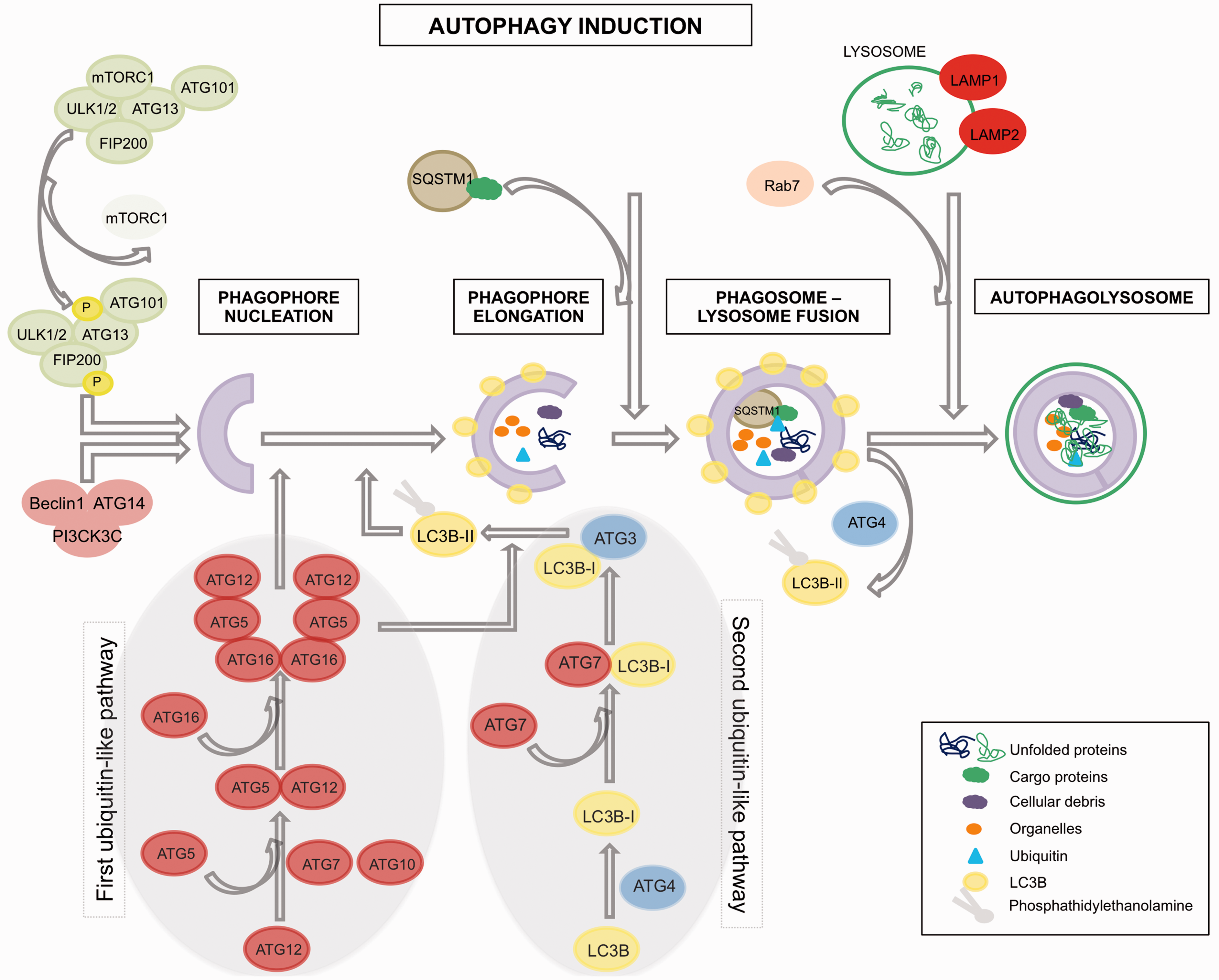
Although autophagy has been considered for long time as an unspecific process for degradation of components of the cytoplasm, recent evidence has revealed that autophagy pathways can differentiate selective load for delivery to the lysosome.21,22 This is guaranteed by the aide of an assistant (p62/SQSTM1), which contains a LC3-interaction domain and an ubiquitin-binding domain, which allows the incorporation of p62 into the autophagosome. p62/SQSTM1 is required for the aggregation of polyubiquitinilated proteins followed by their autophagic degradation. 22 During this process, p62 is also efficiently degraded in the autophagosomes, in contrast to autophagy-deficient cells in which p62 is accumulated intracellularly rather than being reduced. Therefore, it can be said that low levels of p62/SQSTM1 correlate with presence of autophagic activity. 23
Autophagy in atherosclerosis
Autophagy modifier biomarkers in atherosclerotic cells and/or plaque.
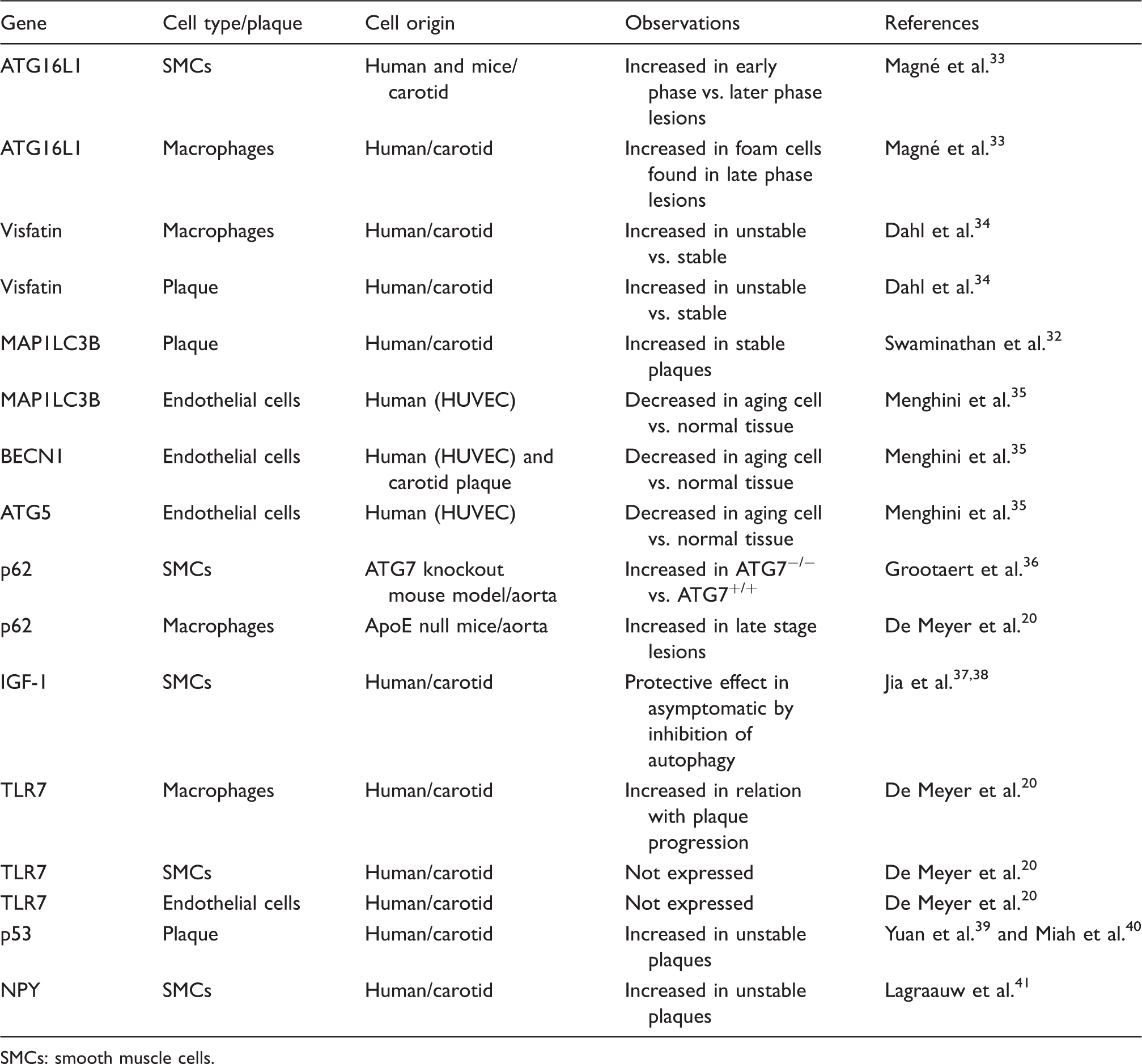
SMCs: smooth muscle cells.
Role of autophagy in atherosclerotic cell types
Autophagic markers have been identified in the main cell types in atherosclerotic plaques from animals with experimental atherosclerosis (i.e. macrophages, endothelial cells (ECs) and SMCs).26,42
Macrophages are known to play an important role in atherosclerosis. At early stages of atherosclerosis, macrophages are involved in the clearance of cholesterol deposits in vascular tissue. Then, when lipids continue to accumulate inside the cells, macrophages are converted in what is called foam cells. However, macrophage phenotype heterogeneity has to be taken into account for the complete understanding of their clinical significance in atherosclerosis. Various macrophage phenotypes present in human atherosclerotic plaques have been recently reviewed
43
and, to the complexity of distinct subpopulations present in the different developmental stages of the plaque, is to be added the specific distribution of subtypes populating the different zones of atheroma plaque. At any rate, macrophage autophagy is known to play a role in atherosclerosis.
44
Liao et al.
27
reported that inhibition of autophagy in macrophages induced plaque necrosis and destabilization in mice. Also, activation of autophagy in macrophages through mTOR inhibition was reported to lead to stabilization of atherosclerotic plaque in mice.
45
These data support also the protective role of autophagic activity in macrophages in atherosclerosis (Figure 2). Macrophage subtype named Mox, presented in murine atherosclerosis different patterns of gene expression than other subtypes (M1, classically classified as pro-inflammatory and M2, anti-inflammatory) including redox-regulated genes; furthermore, Mox showed decreased autophagic activity compared with M1 and M2 in mice.
46
Since the Mox type appeared to represent only 30% of the macrophages present in the advanced plaque, caution is needed when interpreting the contribution of macrophage autophagy to atherosclerosis.
Effects of autophagy on plaque stability.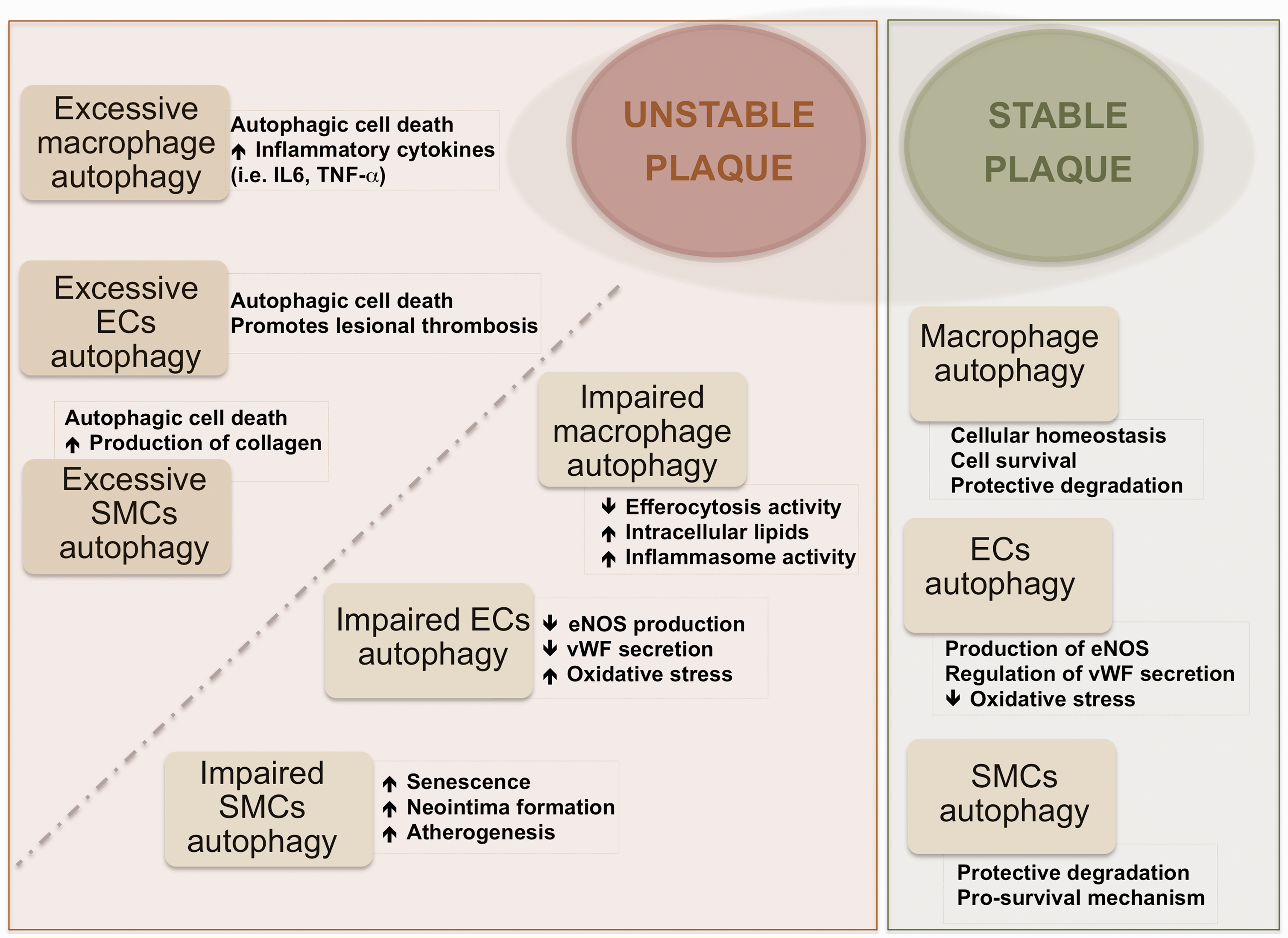
Advanced atherosclerotic plaques have been associated basically with high content of macrophages, whereas unstability of plaques would correlate more strongly with the distinct macrophage phenotypes and their ability to change phenotype than with the quantity of cells. It is indeed likely that in the same plaque different macrophage phenotypes with distinct biological features co-exist, and these may exert contrasting, or even opposing, effects (i.e. macrophages showing autophagic activity or showing deficient autophagy). The overall balance of all the factors playing some role might be determinant of the stability/instability of atherosclerotic plaque. Moore et al. 47 reported the capacity of macrophages to promote plaque regression by changing phenotype in atherosclerosis via administration of the M2-driving cytokine IL-13 to mice. At the same time, Tian et al. 45 reported that induction of autophagy drives macrophages towards the M2 phenotype. It is known that macrophages with impaired autophagy cannot be cleared by efferocytosis; in contrast, these macrophages become apoptotic, and the inflammasome pathway may be activated leading to lesion progression (Figure 2). 48 This thus raises the question whether the observed plaque regression is related with the role of autophagic activity in those specific macrophages.
SMCs are the main component of the vascular system and can experience phenotypic change during atherosclerosis development (i.e. synthetic, macrophage-like or osteochondrogenic phenotypes). 48 SMCs are not passive bystanders but are considered crucial player in atherosclerotic processes. Autophagy exerts a critical role in atherosclerosis by regulating vascular SMC phenotype. 29 Autophagy within normal levels of activity in SMCs is associated with survival and plaque stability while excessive autophagic activity might lead to SMC death and plaque destabilization as reported in different species.31,49 At early stages of atherosclerosis, exposure of vascular SMCs to relatively modest concentrations of oxLDL was shown to increase autophagy that acts as protective mechanism, whereas exposure to high concentrations, which can happen in later stages of atherosclerosis, resulted in a reduction of protective type of autophagy and augmentation of autophagic-mediated cell death in cells from mice aortas. 50 In addition, recently, several studies provided evidence for distinct modes of autophagy induction in vascular SMCs leading to different outcomes. For instance, platelet-derived growth factor, known to be secreted during vascular injury by several cell types, protects against cellular death through activation of autophagy, 29 and other factors, such as osteopontin, have been shown to induce autophagic-driven death in vascular cells from human aortas. 51 Insulin-like growth factor-1 (IGF-1) promotes cell survival by inhibition of autophagy in SMCs isolated from carotid plaques (Table 1) 37 ; however, this effect was only observed in SMCs from asymptomatic plaques. 38 Recently, Grootaert et al. 36 observed detrimental effects of autophagy inhibition in vascular SMCs isolated from atg7 knockout animal model (impaired autophagy), which resulted in higher senescence, neointima formation and atherogenesis and as well as accumulation of SQSTM1.
Vascular ECs are one of the most active cells in the human body, and functional changes in these cells are as well correlated with cerebrovascular diseases. It is known that autophagy plays a role in maintaining a correct endothelial function. 48 Autophagy in ECs induces eNOS expression, and consequently NO availability would reduce oxidative stress in human cells. 52 Also, autophagy regulates the secretion of endothelial von Willebrand factor in human ECs, which is known to play a role in blood coagulation. 53 Recent studies have demonstrated that autophagic activity is present also in ECs in atherosclerotic plaques, as implied by high expression levels of the autophagic marker ATG16L1.33,54 Oxidative stress, which occurs in the atherosclerotic environment, has been observed to trigger autophagy also in human endothelial aortic cells. 55 Similarly to vascular SMCs and macrophages, vascular ECs exposed to excessive autophagic activity can suffer autophagic-mediated death provoking a disruption in the stability of the plaque. 8 Dahl et al. 34 have found visfatin expression levels to be increased in instable symptomatic human carotid plaques. Since visfatin induces the PI3K/Akt/mTOR pathway (meaning inhibiting autophagy), 56 higher visfatin levels observed in vascular ECs 57 could be associated with a decreased autophagic activity.
In summary, evidence is available that the induction of autophagy activity protects against cellular damage in the main cell types involved in atherosclerotic disease (Figure 2). 30 The autophagy-associated biomarkers identified so far as differently expressed in unstable and stable plaques are summarized in Table 1.
Conclusions
Recently, autophagy has been receiving considerable attention since its associated pathways may constitute potential targets for therapeutical interference in atherosclerosis disease. Several drugs have been reported that act to either activate or inhibit autophagy activity.58,59 Particularly, mTOR inhibitors (i.e. rapamycin and derivatives) have been tested as potential plaque stabilizing drugs in animal models. For example, Verheye et al. 60 found that rapamycin analogue everolimus reduced macrophage content in cholesterol-treated rabbits while SMCs remained unaffected. However, while several works have hallmarked autophagy as a potential strategic therapeutical target,59,60 others have pointed to the harmful effects that stimulation of autophagy may exert on pathogenetic processes.8,20,31 In addition, caution is needed when evaluating the effects of specific drugs used to induce/inhibit autophagy activity (for instance, imiquimod exerts autophagy-independent effects). 20 Cell death may be triggered by excessive autophagic activity, such as that observed in macrophages, SMCs and ECs in specific circumstances. Could the unwanted dysfunctional autophagy be targeted to become active again? Several reports have observed that autophagic inducers through mTOR inhibition are capable of reducing plaque progression in advanced atherosclerosis. 61
To gain a more coherent understanding of the role of autophagy in stable or unstable carotid atherosclerotic plaque, it is essential to take into consideration the following variables: (1) the diverse cell phenotypes existing in the plaque at different stages; (2) the specific morphology and stage of the plaque (stable/unstable); (3) the autophagy activity level (inexistent/regular/excessive) found in the distinct phenotypes and in different plaque types; (4) the specific autophagy pathway/s involved in each specific cell type or phenotype, type of plaque (i.e. mTOR dependent or independent).
Footnotes
Acknowledgements
None.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Guarantor
IA.
Contributorship
IA researched literature and conceived the report. IA wrote the first draft of the manuscript. KV was involved in the first draft reviewing. IA, HG, KV and MMF reviewed and approved the final version of the manuscript.