
Abstract
Dermatofibrosarcoma protuberans (DFSP) is an extremely rare cutaneous tumor in children, marked by local aggressiveness, slow growth, high recurrence rate, and low metastatic potential. Its prevalence is often underestimated in children due to its slow growth and frequent misdiagnosis. Diagnosing DFSP can be challenging due to nonspecific symptoms. While most cases present as nodular lesions on the trunk or proximal extremities, some lesions, such as atrophic plaques or sclerotic nodular plaques, can mimic vascular malformations and confuse clinicians. Histologic and immunohistochemical studies are essential for definitive diagnosis. The treatment of choice is complete surgical resection with wide margins to reduce the risk of recurrence. We report two pediatric DFSP cases on the trunk, including one mimicking vascular malformations. Both cases had successful 4 cm margin resections, with no recurrences observed after 6 months and 2 years of follow-up, respectively. Continuous surveillance will be maintained for at least 5 years.
Introduction
DFSP is an uncommon cutaneous tumor in children, characterized as a locally infiltrative dermal and subcutaneous fibroblastic tumor of intermediate malignancy, with limited metastatic potential but high local recurrence rates. 1 The incidence of this rare tumor is approximately 1 case per 1 000 000 person-years in adults, it is even less common in children, comprising only 6% of cases.2-4
The clinical, histological, and immunohistochemical characteristics of pediatric DFSP are similar to those in adults. 5 Its rarity and the lack of awareness among physicians often leads to misdiagnosis and is managed incorrectly as vascular malformations or other benign lesions, causing diagnostic delays with a median time from tumor onset to diagnosis ranging from 3 to 5 years.6,7
The treatment of choice for DFSP is complete surgical excision with wide local excision (WLE) or Mohs micrographic surgery (MMS) to ensure tumor-free margins and minimize recurrence risk. 8 Achieving clear margins can be challenging, especially in the pediatric population, due to difficulties in diagnosing DFSP at a young age and the tendency for late presentation with large lesions. 9
This study aims to describe DFSP in the pediatric population, including its clinical profile and treatment options.
Case Presentations
Case 1
A 7-year-old African descent girl with no previous medical history presented with a persistent swelling in the left supraclavicular region that had been noticed for 7 months. The parents, concerned about the growth and persistence of the nodule despite minimal trauma, sought medical advice. The child did not exhibit any systemic symptoms. Upon examination, the nodule was found to be firm, smooth, painless, and purplish, measuring about 1.0 cm × 1.0 cm × 0.5 cm. The lesion involved the subcutaneous layer and the overlying skin but was not adherent to the underlying musculature, with no signs of axillary lymphadenopathy.
A marginal excision biopsy was performed (1 cm circumferentially and deep). Histopathological examination revealed a dermohypodermal tumor proliferation with poorly defined borders, displaying a fasciculated architecture and spindle-shaped tumor cells. No significant atypia or mitotic activity was observed (Figure 1). Immunohistochemical staining showed positive and diffuse staining with anti-CD34 antibody (Figure 2). Tumor cells were negative for PS100, AML, Desmine, and H-caldesmon antibodies confirming the diagnosis of DFSP.
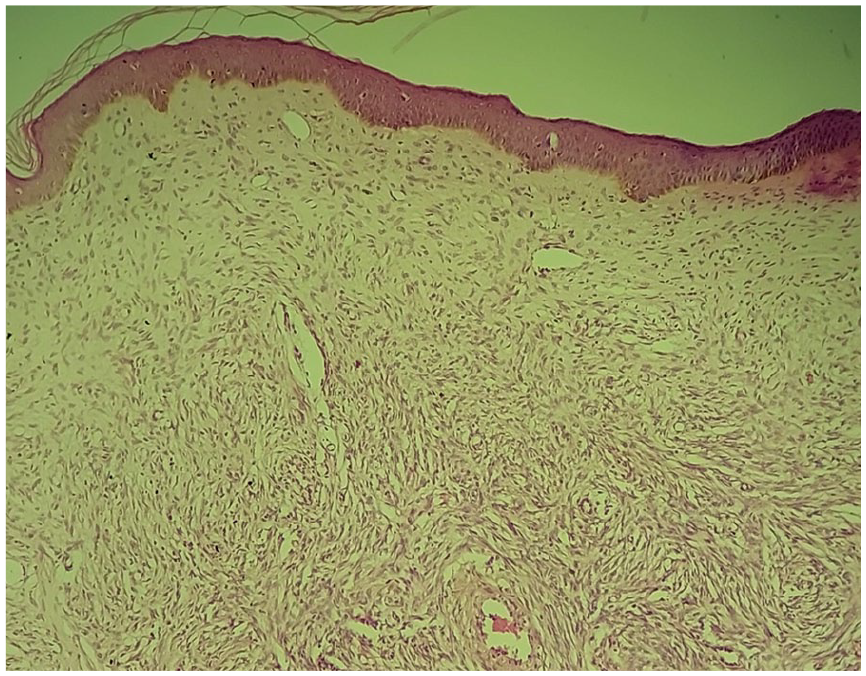
Microscopic findings showing a dermo-hypodermal tumor proliferation with poorly defined borders, exhibiting a fasciculated architecture and spindle-shaped tumor cells (Hematoxylin stain, x40 original magnification).

Photomicrograph showing positive staining of tumor cells with anti-CD34 (Immunostain, x40 original magnification).
Post-diagnosis imaging, including chest X-ray, abdominal ultrasound, and chest computed tomography scan, showed no evidence of metastasis. Following a multidisciplinary team discussion, a second surgical excision was performed with wide local excision (WLE), encompassing a 4 cm radial margin around the biopsy scar, excising the epidermis, dermis, and aponeurosis of the deltoid and trapezius muscles to a depth of 2 cm (Figure 3A, B, and C). The defect was resurfaced with a meshed split-thickness skin graft (Figure 3D). No adjuvant therapy was required, and no recurrence was observed during the 2-year follow-up period.
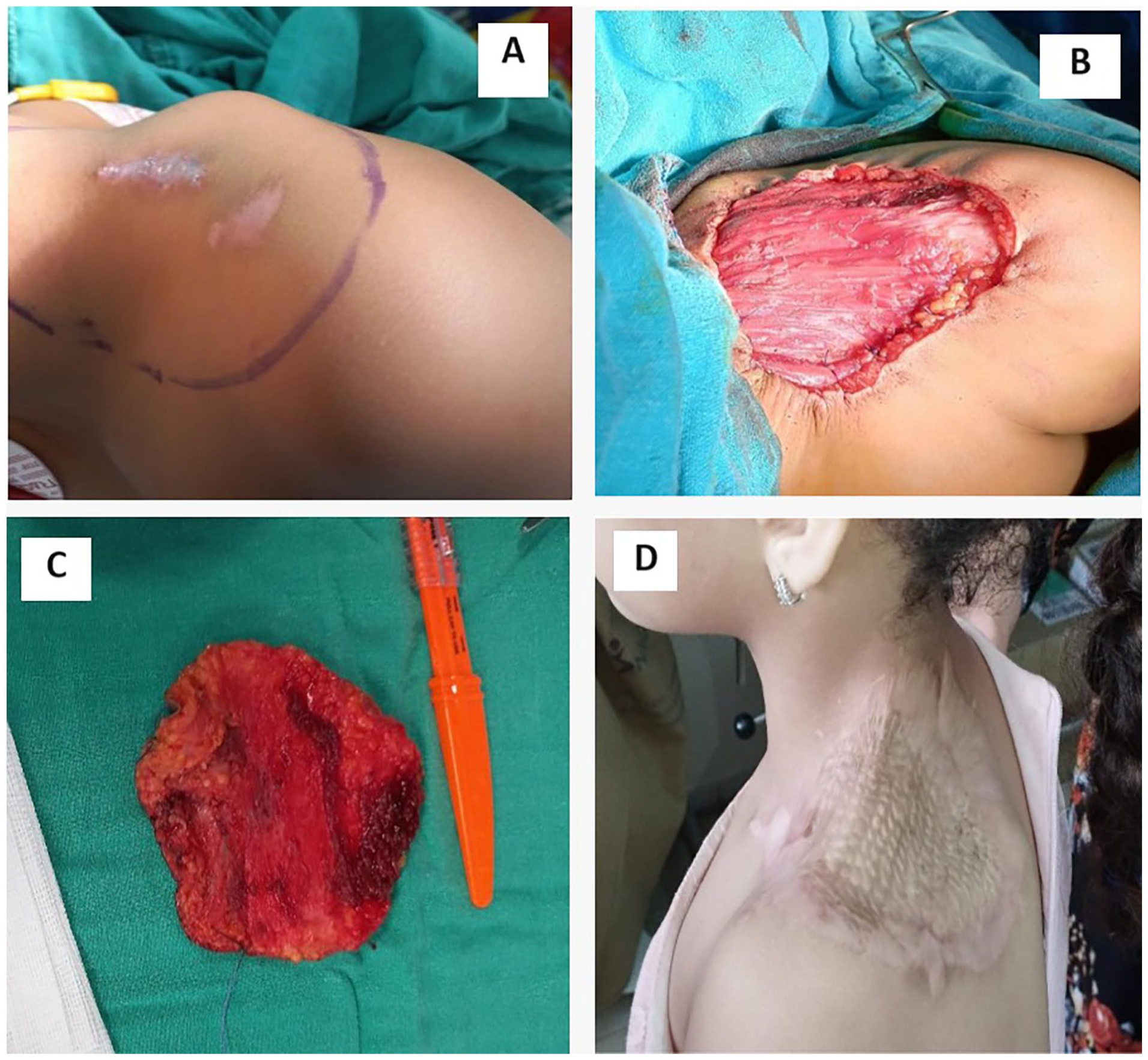
(A) Clinical photograph of biopsy scar before the second surgical excision with preoperative markings. (B) Preoperative picture showing wide local resection (WLE) with 4 cm margins. (C) Image of the surgical specimen after wide excision, 4.0 cm × 4.0 cm × 2 cm. (D) Clinical photograph of the skin grafted at 12 month follow-up after surgery.
Case 2
A 10-year-old African-descent boy, previously healthy, presented with a progressively enlarging, large sclerotic nodular plaque on the right supraclavicular area, measuring 3.2 cm × 1.2 cm × 1.5 cm, noted for over 2 years (Figure 4A). The lesion had become pruritic, prompting medical consultation.
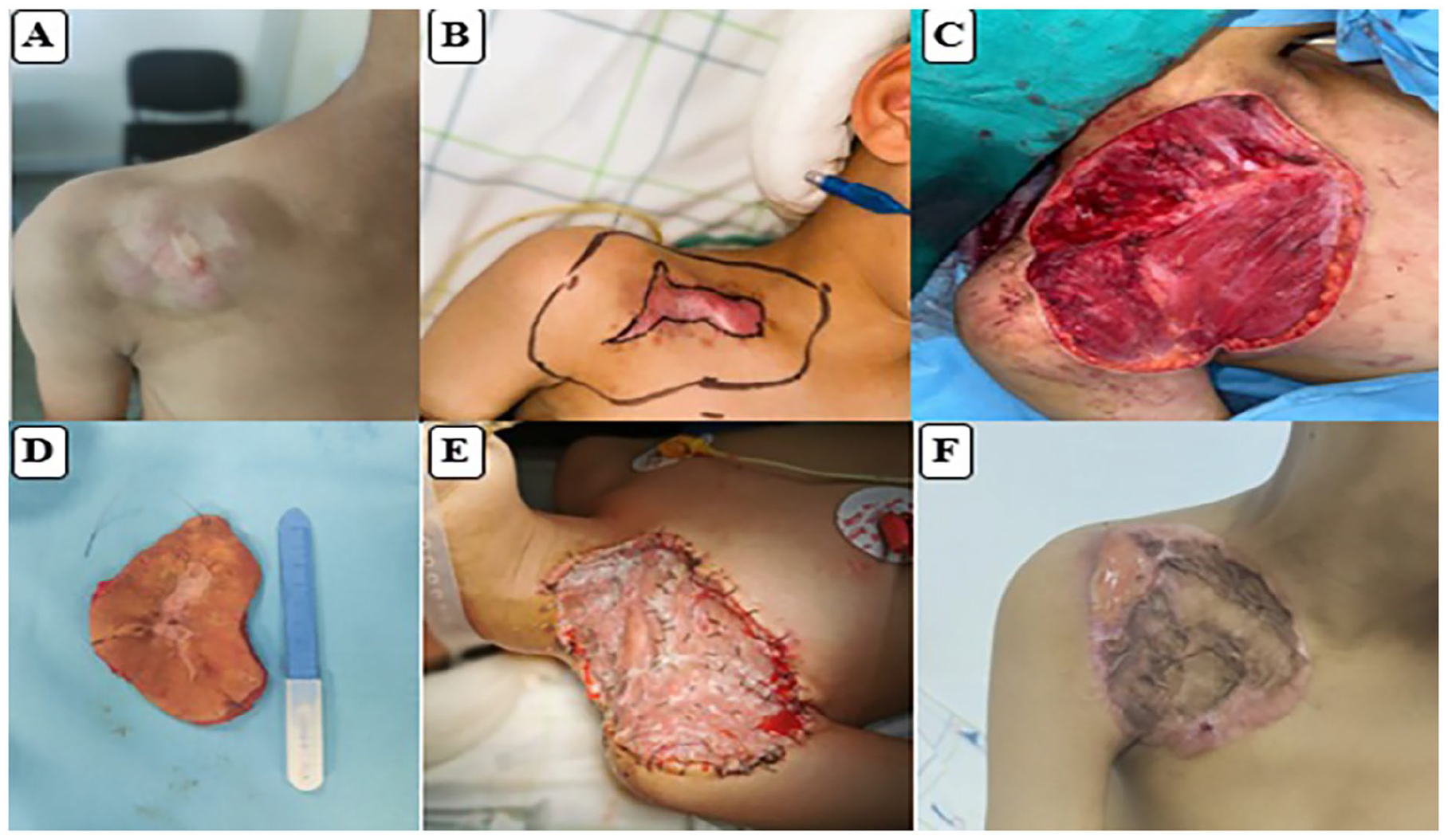
(A) Clinical photograph of the 5.2 cm × 1.4 cm × 1.7 cm large sclerotic nodular plaque skin mass on the right supraclavicular. (B) preoperative markings. (C) Preoperative picture showing wide local resection (WLE) with 4.0 cm margins. (D) Image of the surgical specimen after wide excision 4.0 cm × 5.0 cm × 2.0 cm. (E and F) Clinical photograph of the skin grafted on postoperative and at 1-month after surgery respectively.
The ultrasound revealed a well-circumscribed, heterogeneous hyperechoic subcutaneous mass with hypoechoic areas and visible vessels, communicating with a branch of the right subclavian artery, measuring 5.2 cm × 1.4 cm × 1.7 cm. Magnetic resonance imaging (MRI) indicated a hypervascular, hypointense T1-weighted, hyperintense T2-weighted subcutaneous mass connected to the superficial branch of the right subclavian artery and extending toward the deltoid muscle’s aponeurosis, initially suspected to be an arteriovenous malformation.
Despite 4 months of medical treatment (Prednisone), there was no improvement, and after 8 months, the patient was referred to the vascular surgery department and underwent surgical resection of the mass. Histopathological examination revealed spindle cell proliferation with a storiform pattern and myxoid differentiation, crossing the surgical margins. Tumor cells showed eosinophilic and abundant cytoplasm; nuclei were monomorphic and ovoid to elongated with variable low mitotic activity (Figure 5). Tumor cells infiltrated and expanded fibrous septa and showed interdigitation among lobules of fat. A panel of immunostains was carried out and showed positive staining with anti-CD34 antibody (Figure 6). Tumor cells were negative for EMA, PS100, AML, Desmine, and H-caldesmon antibodies, with a Ki67 proliferation index of approximately 10%. These features were consistent with a diagnosis of DFSP.

Microscopic findings showing a tumor proliferation of spindle cells with a storiform pattern (Hematoxylin stain, x100 original magnification).
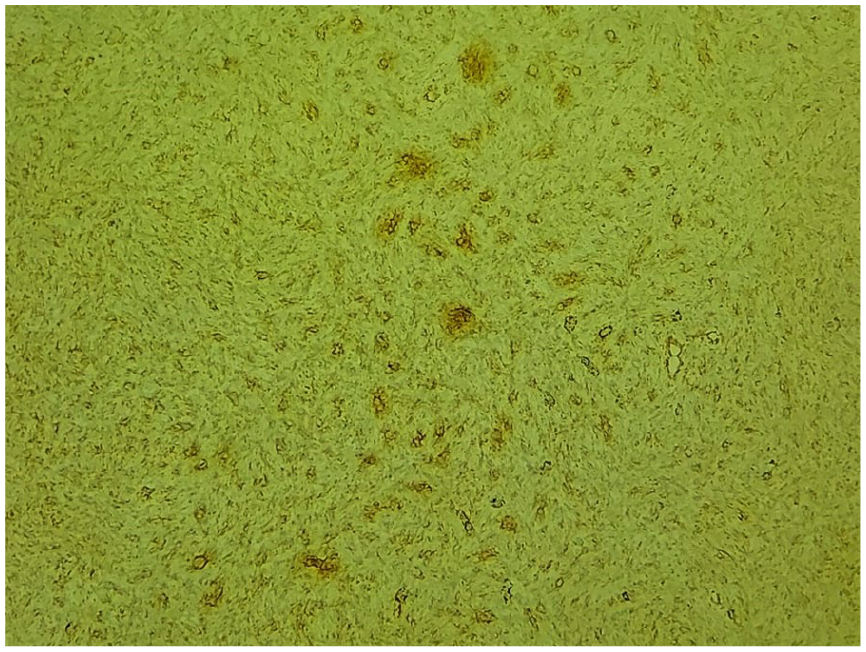
Photomicrograph showing positive staining of tumor cells with anti-CD34 (Immunostain, x100 original magnification).
Post-diagnosis imaging, including chest X-ray, abdominal ultrasound, and chest CT, showed no metastasis. Following a multidisciplinary team meeting, a second WLE was performed, achieving 4.0 cm × 5.0 cm × 2.0 cm surgical margins (Figure 4B, C, and D), followed by a meshed split-thickness skin graft (Figure 4E and F). No adjuvant therapies were necessary. Follow-up at 8 months showed no signs of recurrence.
Ethical Approval and Informed Consent
Ethical approval was not required for this case report. Written informed consent was obtained from the patients and their parents for publication.
Discussion
DFSP, first described by Hoffman and further elaborated by Darier and Ferrand, is classified by the World Health Organization (WHO) as a low-grade sarcoma.10,11 It’s a rare mesenchymal tumor that, although primarily affecting adults, can present unique diagnostic and therapeutic challenges in the pediatric population.6,7 Its incidence in children is extremely low, accounting for only 6% of all DFSP cases. 5 The incidence in adults is higher among women and the black population, although the gender predilection is less clear in children.2,5,7
The pathogenesis of DFSP involves a chromosomal translocation, specifically t(17;22)(q22;q13), resulting in the formation of the COL1A1-PDGFB fusion gene, which drives tumor growth. This molecular abnormality can be detected using fluorescence in situ hybridization (FISH), aiding in diagnosis and validating the use of targeted molecular therapies in certain cases. 3 No known predisposing factors for DFSP have been identified, but it has been associated with a history of trauma, and can develop in old burn wounds, surgical scars, and sites of multiple immunizations.11,12
DFSP typically manifests as a slow-growing, firm, and nodular lesion that can be asymptomatic for years. 6 It can appear as a single brownish or purplish plaque-like area of cutaneous thickening or as a small raised nipple-like projection. 14 In pediatric patients, the tumor often presents on the legs and acral regions, although congenital forms may follow the adult pattern, appearing on the trunk and proximal limbs.2,3,9,12 The progression from a small, indolent lesion to a more prominent multinodular mass can take several years, contributing to diagnostic delays. 14 Tumor sizes vary widely, ranging from 0.5 to over 10 cm in diameter, with a mean size of 2–3.5 cm. 15 Early-stage lesions may be mistaken for benign conditions, delaying appropriate treatment. 16 Patients may initially perceive the primary lesion as a benign keloid or scar. However, the tumor is progressive, extensive, and often unresponsive to usual local symptomatic treatment. Patients typically seek medical attention when the appearance of the tumor changes or when symptoms such as ulceration, infection, or bleeding develop. 14 As observed in our cases where the diagnosis was only confirmed after significant tumor progression.
Due to its heterogeneous presentation, DFSP is frequently misdiagnosed, leading to delays in diagnosis. The median diagnostic delay for DFSP is approximately 4 years. 16 Misdiagnoses are more frequently made by primary care clinicians (75%) and dermatologists (33%), though other types of physicians may also misdiagnose DFSP. 7 DFSP in children often goes unrecognized due to its rarity and the frequent misdiagnosis as benign lesions such as cysts, lipomas, or vascular malformations.6,17
Accurate diagnosis of DFSP in children involves a thorough approach that includes a detailed medical history, physical examination, and a generous biopsy (punch or excisional) for histopathological and immunohistochemical evaluation. DFSP is characterized by spindle cell proliferation with a storiform pattern and CD34 positivity. 1 Molecular diagnostic techniques, such as fluorescence in situ hybridization (FISH), can detect the characteristic COL1A1-PDGFB fusion gene, further confirming the diagnosis and providing insight into targeted therapy options. 3 Imaging studies, including ultrasound, CT, MRI, and PET/CT, are essential for evaluating the extent of the tumor and planning surgical intervention.18-20 Both our cases demonstrated the importance of a thorough clinical and histopathological evaluation supported by immunohistochemical analysis showing CD34 positivity, which is crucial for accurate diagnosis.
The imaging features of DFSP are variable and not specific, with High-frequency ultrasound is particularly useful for assessing tumor extent and guiding biopsies, often revealing a hypoechoic or mixed hyperechoic superficial nodular mass with well-defined or irregular margins. The vascularity of DFSP, which indicates malignancy, also varies. 19
Computed tomography (CT) scans can identify a solitary, subcutaneous lobular or nodular structure with soft tissue attenuation and post-contrast enhancement, which is particularly helpful for evaluating distant metastases. 20 Areas within larger tumors (>5 cm) that do not enhance after contrast administration may indicate necrosis and cystic degeneration. PET/CT using 18F-fluorodeoxyglucose (FDG) can be valuable in detecting metastatic disease and monitoring treatment response. 20
Magnetic resonance imaging (MRI) is essential for determining the size, extent, and relationship of DFSP with surrounding structures. Therefore, MRI is recommended for preoperative evaluation, surgical planning, and recurrence monitoring. 21 T1-weighted MRI images typically show well-defined, homogeneous isointense lesions, while T2-weighted images reveal well-defined subcutaneous soft tissue nodules or masses with intermediate-to-marked homogeneous hyperintensity relative to surrounding muscle tissue. 22 Poorly defined irregular margins may be observed in some cases. 23 In our case 2, imaging helped delineate the tumor boundaries and assess the involvement of underlying structures, which is essential for surgical planning.
Macroscopically, DFSP usually appears as a poorly circumscribed, white to yellow, soft-tissue mass with a solid, fish flesh-like texture. Larger tumors may exhibit hemorrhagic or cystic changes. 25 Histologically, DFSP arises from fibroblasts in the dermis or subcutaneous tissues. 24 Early-stage DFSP features loosely scattered spindle cells in the upper dermis, progressing to monomorphous spindle cells arranged in a storiform pattern in later stages. Immunohistochemical staining typically shows CD34 positivity in the spindle cells, with negativity for other markers such as protein S100, Factor XIIIa, alpha-smooth muscle actin, and melanA. 1 DFSP has several histological variants, including myxoid, pigmented, giant cell, giant cell fibroblastoma, granular cell, sclerotic, and fibrosarcomatous (FS) components. 11 The FS component, present in 10% to 20% of cases, is an intermediate-grade sarcoma with a higher risk of local recurrence and metastasis (5-15%).1,25 Classic DFSP, which lacks the FS component and accounts for 80% to 90% of cases, is considered a low-grade malignancy with a high local recurrence rate but low metastatic potential. 13
The differential diagnosis for DFSP in children includes various benign and malignant tumors, such as vascular malformations, keloids, neurofibromas, dermofibromas, hemangiomas (particularly in children), schwannomas, solitary fibrous tumors, spindle cell lipomas, and melanomas.6,23
The primary treatment for DFSP is surgical resection with wide local excision (WLE) or Mohs micrographic surgery (MMS) to ensure tumor-free margins and minimize recurrence risk.25,26 Achieving clear margins in pediatric patients can be challenging due to the need to balance oncologic control with the preservation of function and cosmesis.11,27 In the presented cases, WLE with sufficient margins followed by skin grafting was successfully performed, with no recurrence observed during follow-up periods of 6 months and 2 years, respectively.
For WLE, a margin of 2 to 4 cm around the tumor is typically excised, ensuring removal of the malignancy and a portion of healthy tissue.25,26 Larger DFSPs may necessitate reconstructive procedures, including local flaps, skin grafts, or myocutaneous flaps. 27 The likelihood of local recurrence largely depends on the thoroughness of the initial excision and the status of the surgical margins. A positive margin generally warrants re-excision.
MMS is an alternative to WLE that involves the stepwise horizontal removal of the tumor with immediate frozen section analysis to confirm tumor-free margins (R0 resection). This method provides precise control over the entire tumor margin while preserving as much healthy tissue as possible.25,26 WLE is typically utilized for DFSP located on the trunk and extremities, where complete excision is often achievable in a single procedure. In contrast, MMS is particularly suitable for DFSP in cosmetically and functionally sensitive areas (head and neck, face, genitalia, and toes) to minimize tissue loss and avoid procedures such as amputation. 27
Multiple studies have shown that MMS significantly reduces the risk of recurrence compared to WLE. 26 For instance, a meta-analysis of 684 patients reported recurrence rates of 9.10% with WLE and 2.72% with MMS over 5.32 years. 27 Additionally, data from the Mayo Clinic revealed a recurrence rate of 30.8% with WLE, in stark contrast to 3.0% with MMS. Furthermore, MMS resulted in primary closure in 73% of cases, whereas WLE often required flaps, grafts, and other closure techniques in 52% of cases. 26
While surgical resection remains the cornerstone of DFSP treatment, adjuvant therapies such as radiotherapy and targeted molecular therapies may be considered in cases where complete surgical excision is challenging or in instances of metastasis. 28 In adults, targeted therapy with tyrosine kinase inhibitors like imatinib mesylate has shown efficacy in unresectable, recurrent, or metastatic DFSP.2,9 Radiotherapy is an option, particularly when surgical margins are positive or close, and re-excision is not feasible. A multidisciplinary systematic review conducted by Fionda et al highlighted the role of postoperative radiotherapy in managing DFSP, concluding that radiotherapy could be beneficial in reducing recurrence rates, especially in cases where achieving clear surgical margins is challenging. 28 However, the use of such therapies in pediatric patients is not well-established and requires further investigation.2,9,14
The prognosis for pediatric DFSP is generally favorable, with 15-year and 30-year overall survival rates of 98% and 97%, respectively, comparable to adult survival rates.1,2,6 Clinical follow-up is crucial every 6 to 12 months, especially in the first 3 to 5 years post-surgery, to monitor for recurrence. Regular evaluations, including physical examination and imaging studies, are recommended to detect any signs of recurrence early. 2 Any abnormal healing or new lesion development should prompt an immediate biopsy to confirm or rule out recurrence.
Conclusions
DFSP in children, while rare, poses significant diagnostic and therapeutic challenges. Early and accurate diagnosis, combined with complete surgical resection is crucial for achieving optimal outcomes. The presented cases underscore the importance of considering DFSP in the differential diagnosis of persistent cutaneous lesions in pediatric patients and highlight the necessity of a multidisciplinary approach in managing this rare tumor. Close clinical follow-up is essential to ensure early detection and management of recurrence. Further studies are needed to establish clear guidelines for managing pediatric DFSP, including the role of adjuvant therapies.
Footnotes
Acknowledgements
We would like to acknowledge the support of the medical and surgical teams at the institutions involved in the care of these patients. We also thank the patients and their families for their cooperation and consent to share their cases.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.