
Abstract
Mevalonate kinase deficiency (MKD) is a rare hereditary autoinflammatory disease, with a widely variable clinical spectrum. It is characterized by febrile recurrent episodes and systemic inflammation. Data on therapeutic options for MKD are still limited and remain unknown in our country. We report Moroccan cases with MKD referred in our unit and treated with Anakinra, an interleukin-1 receptor antagonist. Through this study, we evaluate the efficacy of this bioagent, in our 2 MKD patients, in whom Anakinra has shown a complete clinical remission, with a remaining mild inflammation for one case, and normalization of growth with rare episodes of cervical adenopathies for the second case. Our experience provides an additional argument supporting the efficacy of Anakinra treatment, demonstrated previously but still lacks of objective data.
Introduction
Mevalonate kinase deficiency (MKD) is a rare autosomal recessive autoinflammatory disease (AID), resulting by mutations in the encoding gene for mevalonate kinase (MVK), an enzyme involved in cholesterol and non-sterol isoprenoids biosynthesis pathway. 1 In recent years, research into the pathogenesis for MKD has shown that Interleukine 1 (IL1), a pro-inflammatory cytokine, plays a major role, leading to the hypothesis that an anti-IL1 drug could be a promising therapeutic choice. 2 In this paper, we report two MKD patients treated successfully with Anakinra.
Case Reports
Written informed consent was obtained from the patients’ legally authorized representatives prior to their inclusion in this report.
Case 1
D., 19 years old with normal cognitive development, was born in 2003 from consanguineous parents, has a surgery for an appendectomy at the age of 4. Since 18 months old, the patient presented recurrent febrile episodes, triggered by vaccines/infections and characterized by a mean fever of 38.5°C, polyarthralgias, abdominal pain, diarrhea and vomiting lasting near 4 days and occurring more than twice a month. At 5 years old, she was hospitalized in our department for recurrent fevers, where the examination showed arthritis of the left knee and splenomegaly with 3.5 cm overflow. Laboratory results showed high inflammatory markers with an erythrocyte sedimentation rate (ESR) of 47.2 mm at the first hour, a C-reactive protein (CRP) of 60.8 mg/l and an Amyloid A serum protein (AAS) at 20 mg/l. The immunoglobulin assay showed increased levels of IgA, IgM and IgG. The immunological tests showed a positive rheumatoid factor but the antinuclear and anti-DNA antibodies were negative.
The patient was considerate juvenile idiopathic arthritis and treated with corticosteroid at a dose of 5 mg/day (0.3 mg/kg/day), methotrexate at 12.5 mg/week (0.9 mg/m2/dose) and non-steroid anti-inflammatory drugs (NSAIDs) on demand without any improvement. Moreover, side effects of corticosteroids were noted such as hyperpilosity and cushingoid fasciae (Figure 1). The persistence of joint involvement and abdominal pain has referred the diagnosis to Familial Mediterranean Fever (FMF) which was excluded, first genetically by the absence of mutations in the exon 10 MEFV gene and secondly by the failure of colchicine treatment.
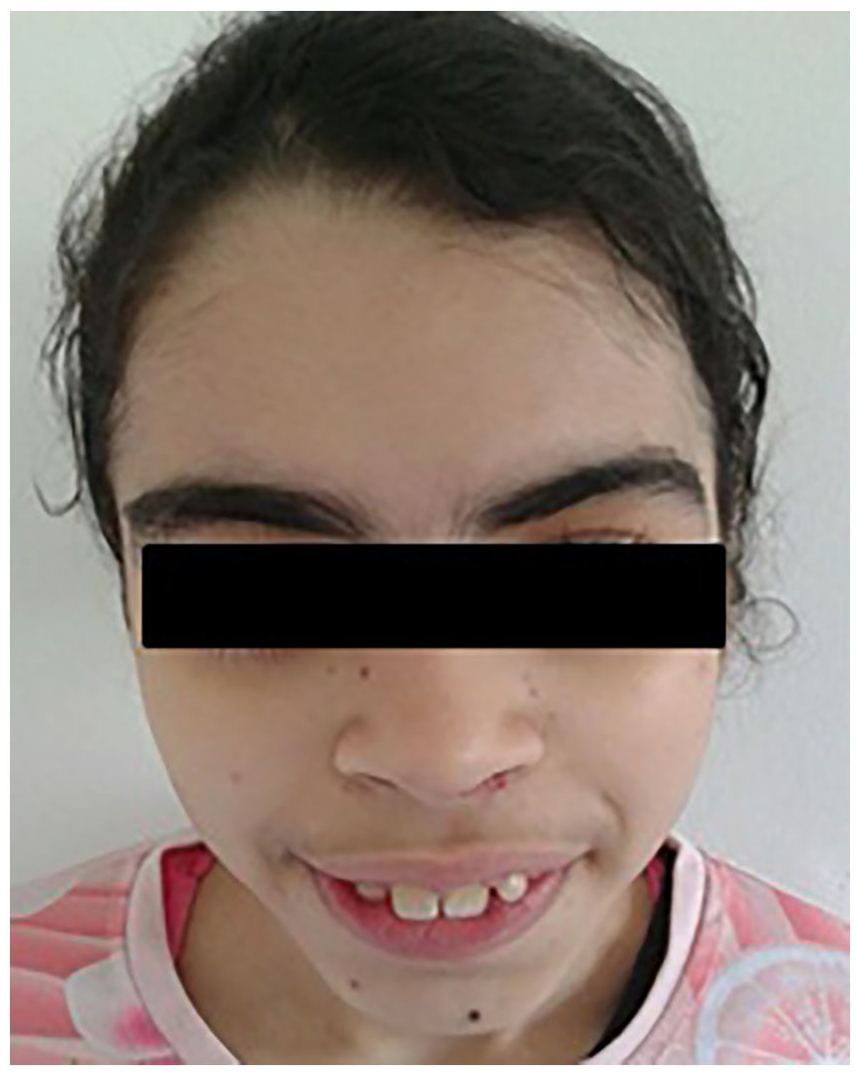
Patient 1 particular fasciae.
The evolution was marked by the recurrence of febrile episodes with persistent painful abdominal attacks, the onset of a splenic overflow, an urticarial rash, a major mouth aphthosis, and extended arthritis to both knees and elbows with increased inflammatory markers over a period of 3 years, triggered by infections. Furthermore, the urinary mevalonic acid assay was increased at 66.7 mmol/mol creatinine during febrile episodes (for a normal value lower than 1.3) leading to the diagnosis of MKD. The diagnosis delay was 10 years.
The patient was treated with Anakinra at a high initial dose of 5 mg/kg/d because of the severity of the disease, reduced to 2 mg/kg/d administrated 1 day out of 2 with a spectacular clinical evolution marked by the absence of all clinical signs (febrile episodes, skin rash and articular or digestive signs with regression of the splenomegaly), but was keeping persistently high inflammatory markers with ESR at an average of 54 mm 1st hour.
Case 2
S., 4 years old, born in 2018, from non-consanguineous parents who report a similar case in a nephew on the mother’s relative (Figure 2). The onset of symptoms was at the age of 5 months following his combination vaccine Penta 3 (DTaP-IPV-Hib) containing tetanus, diphtheria, acelullar pertussis, inactivated poliomyelitis virus, conjugated-Hib polysaccharide antigens, associated to viral hepatitis type B and invasive pneumococcal vaccines. He started his disease by recurrent fevers impacting his growth and psychomotor development. He was admitted at the age of 9 months after ruling out an immunodeficiency in the Clinical imuunolgy and infectious diseases department by a normal immunoglobulin dosing, T lymphocytes subpopulation and negative DHR and HIV tests. Inflammatory attacks were characterized by febrile episodes of 39 to 40°C, vomiting, persistent painful cervical adenopathies, oral aphthosis, temporary skin rash, and pallor, occurring 1 to 2 times a month during 3 to 4 days. Clinical examination showed splenomegaly with a 4.5 cm overflow, and hepatomegaly of the left sided liver.

Patient 2 family tree.
The biological tests revealed an inflammatory syndrome with a mean CRP of 32 mg/l and ESR of 35 mm at the first hour and an AAS level of 96 mg/l. The metabolic screening showed a high level of urinary mevalonic acid at 435.9 mmol/mol creatinine during febrile crisis, thus confirming an attenuated form of mevalonic aciduria with a diagnosis delay of 2 months without any genetic confirmation or enzymatic activity assay performed.
The patient was treated initially with a 2 mg/kg/d (25 mg/d) dose of Anakinra, progressively increased to 6.25 mg/kg/d (75 mg/d) allowing to a decreasing of the biological markers, to the remission of the clinical signs and the normalization of the growth rate (Figure 3) but an occasional occurrence of the febrile episodes and cervical adenopathies with a current decline of 4 years (29.07.2022).
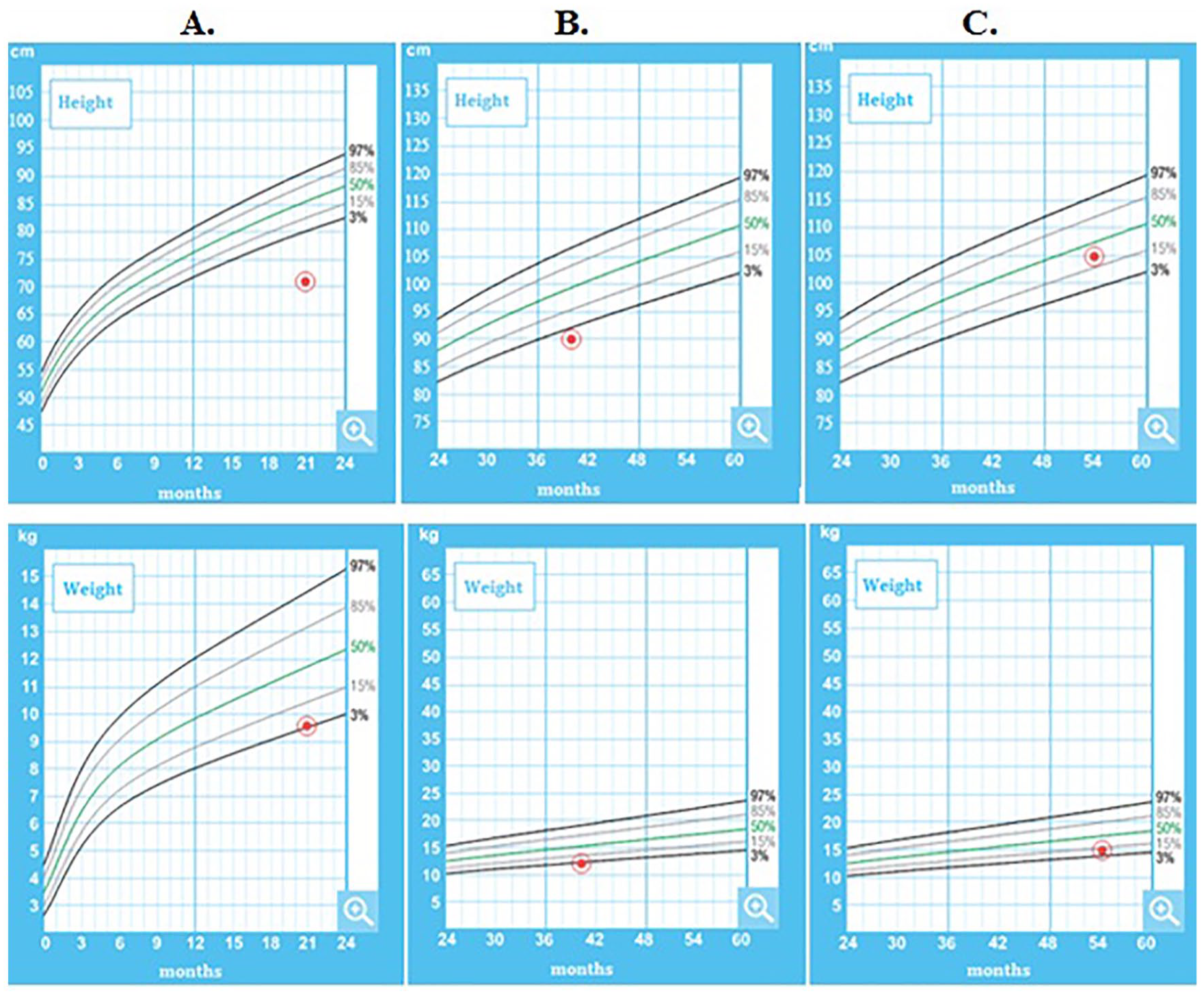
Patient 2 growth curve.
Discussion
MKD is an autosomal recessive disease resulting from “loss of function” mutations in the MVK gene, of which the most frequently reported is the founder mutation “V377I.”1,3 It is a rare worldwide disease affecting more than10 000 patients in both sexes equally. The exact prevalence of MKD is still unknown considering a certain number of non-included, undiagnosed and/or unreported cases especially in the Arab population with high consanguinity. 4
Due to the altered activity of the encoded mevalonate kinase, the lack of isoprenoids, in particular “geranylgeranyl-phosphate,” leads to the inactivation of RhoA (a protein required to silence pyrin), resulting in the activation of pyrin-inflammasome “caspase-1” through Rac1 signaling pathway and subsequently, the constitutive release of inflammatory cytokines such as IL-1β, IL-6, and Tumor Necrosis Factor α (TNFα).5,6 Moreover, defective isoprenylation of the small GTPase may also contribute to inflammation in MKD, by leading to enhanced activation of IL-1β through NFκB transcription pathway. 3 The cytokines interaction in MKD is not yet fully understood, and the exact mechanism by which the metabolic defect gives rise to the clinical and immunological features of the disease remains partly understood. 7
The clinical spectrum of MKD ranges from attenuated forms of hyperimmunoglobulinemia D syndrom (HIDS, OMIM #260920) in case of partial MVK deficiency (1.8%-28% of normal) suspected in case 1 to lethal forms of mevalonic aciduria (MA, OMIM #610377) in case of a complete absence of MVK activity, 8 with a mild phenotype suspected in case 2 based on clinical signs. Symptoms appear particularly in the first year of life in 90% of patients, as found in case 2. They may be triggered by stress, infection or vaccination and are characterized by episodes of high fever (>39°C) lasting 3 to 7 days and occurring every 4 to 8 weeks. They are associated with painful cervical adenopathies (95%), abdominal pain (70%) with splenomegaly, vomiting (55%) and diarrhea (80%). Arthralgia (80%) or arthritis (70%), skin rash (80%), mouth ulcers (50%) and headache (50%) are also frequently associated as found in our patients.3,9 The clinical and biological signs return to normal between the episodes, and the disease is very rarely complicated by AA amyloidosis. 5
According to well-defined criteria of the European Registry EUROFEVER, the diagnosis is based generally on recurrent febrile episodes stereotyped more than 3 times in a year, spaced apart by symptom-free intervals and out of any context of infection, and particularly, on the presence of elevated biological inflammatory markers during crisis with immunological and genetic analysis showing an homozygous or heterozygous composite mutation in the MVK gene, or a heterozygous mutation in the presence of an increased excretion of urinary mevalonic acid during febrile episodes or a low level in the activity of the mevalonate kinase in fibroblasts.1,10 Elevation of IgD serum level is not necessary because non-specific. 9 The diagnosis of our patients was based on recurrent fevers, inflammatory syndromes, common symptoms of MKD, increased urinary mevalonic acid in the absence of the enzymatic activity and genetic research in Morocco, and considering the notion of consanguinity or similar family case.
To date, data on the various treatments for MKD are limited and mainly based on clinical cases or small series, more often in pediatric patients and within limited follow-up periods 11 A large number of drugs have been used including colchicine, azathioprine, cyclosporine and methotrexate with low or no efficacy.4,12 Moreover, many treatments have been tried to prevent and treat febrile episodes such as the on-demand use of NSAIDs, Paracetamol and corticosteroids with partial improvement by only reducing the duration of febrile episodes. 5 Since the understanding of the role of the inflammasome and the overproduction of IL-1β in MKD, biologic agents targeting IL-1β have been adopted including “Anakinra,” which is a recombinant form of the human IL-1 receptor antagonist that acts by inhibiting competitively the binding of IL-1 with its type 1 receptor. 13 Anakinra could be used daily or during febrile attacks but maintained treatment decreases serum amyloid A level thus prevent amyloidosis. 14 As found in case 1, it is currently approved to be effective for decreasing the frequency and severity of fever attacks and should be considered to limit corticosteroid exposure. 5 It has been shown that it is more effective if administrated when the first signs of MKD appear to avoid psychomotor or growth delay, starting with a dose of 2 mg/kg/day, and may increase to 5 to 8 mg/kg/day as shown in case 2. 1 Among the studies reporting Anakinra-treated patients, 11% to 30% had a complete response at mid-term follow-up, whereas 78% achieved a partial one. At long-term follow-up, Anakinra showed a 100% complete response. In other observational study, treatment with Anakinra resulted in complete clinical response in 52% and improvement of health conditions in 81% of patients. 15 Furthermore, in a Finnish pediatric experience of 33 patients, 21 children with MKD treated with Anakinra, had a complete or partial/ positive response in 90% of cases, suppressing inflammation with the absence of most symptoms. 12 According to the retrospective data from the international registry that includes both adults and children, more patients responded to Anakinra (24/27, 89%) than to Etanercept (11/17, 65%). 16 Nevertheless, incomplete responses to Anakinra suggest the implication of others cytokines. 14 Table 1 summarizes the response to Anakinra in different case series and case reports, highlighting a good but rather partial response in the majority of cases, as shown in our two patients. Recent evidence from observational studies and clinical trials showed the efficacy of other IL 1-targeted therapies such as Canakinumab in Cryopyrin Associated periodic syndrome “CAPS,” TNF Receptor Associated Periodic Syndrome “TRAPS,” Familial Mediterranean Fever “FMF” and MKD, revealing a good safety profile with minor concerns for tolerability. 12 It is a human monoclonal antibody, alternative to Anakinra and currently approved for auto-inflammatory diseases in several countries in adults, adolescents and children aged at least of 2 years, but not available in Morocco. The clinical use of this bioagent in MKD has been approved with satisfactory results leading to a disappearance or a significant reduction in the severity of MKD features, including fever, lymphadenopathy, abdominal pain and aphthous ulcers.13,17 Whereas Rilonacept, a soluble receptor, is more indicated in CAPS. In a placebo-controlled study, the disease activity score decreased by 84% in patients receiving Rilonacept, compared with those receiving placebo (13%). However, it is not approved in Europe and only available in the United States. 18
Efficacy of Anakinra Treatment in MKD Patients in Different Series and Case Reports.

Currently, activity scores trials of assessment are in process. The AutoInflammatory Disease Activity Index (AIDAI) questionnaire is recommended by the international Single Hub and Access point for pediatric Rheumatology in Europe (SHARE) initiative but it is still under evaluation in order to unify the outcome measures to be objective in the assessment of activity of AIDs. 18
Conclusion
Our report highlights the challenge of making a definitive diagnosis of MKD on the basis of specific clinical symptoms and paraclinical parameters such as urinary mevalonic acid assay in the absence of genetic testing availability, in order to reduce the diagnosis delay and ensure an early therapeutic care. Similar to previous data, treatement with Anakinra could be effective to reduce the severity of attacks and improve the quality of life in patients with MKD. Our cases provide an additional argument for the efficacy of Anakinra in MKD patients in Morocco awaiting for better targeted treatment.
Footnotes
Authors’ Contribution
The PhD student Souali participated to conceptualization and design of this work, and drafted the manuscript. Assistant Professor Sakhi is a permanent pediatric rheumatologist in the unit. Professor Bousfiha is the director of Clinical Immunology, Inflammation and Allergy laboratory. Professor Bouayed conceptualized and designed this study, and critically reviewed the manuscript for important intellectual content. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Written informed consent for publication was obtained from the legally authorized representatives of the patients and held in their hospital records. For studies involving human participants, all procedures performed were approved by the institutional ethics committee of the University Hospital Center Ibn Rochd under the reference 2021/DOEHRSI/31 and file N° 03/21, and conformed with its ethical standards as well as with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.