
Abstract
Holoprosencephaly (HPE) is a rare birth defect that occurs during the first few weeks of pregnancy. It results from a disturbance in the usual signaling pathways required for separation of the embryonic prosencephalon into 2 separate cerebral hemispheres. Classically four subtypes have been recognized: alobar, semilobar, lobar, and middle interhemispheric holoprosencephaly. The cause of HPE is unknown but may include genetic disorders. In most cases of holoprosencephaly, the malformations are so severe that babies die before birth. In less severe cases, babies are born with normal or near-normal brain development and facial deformities that may affect the eyes, nose, and upper lip. We report 2 cases of semilobar holoprosencephaly, diagnosed in Children’s Hospital of Rabat: the first one was a fetus diagnosed by ultrasonogram at 25 weeks of gestation. The second one was a newborn at 15 days of life diagnosed by brain scan.
Introduction
Holoprosencephaly is a rare brain malformation resulting from the incomplete division of the prosencephalon during the third and fourth week of gestation. The result is a single-lobed brain structure. Its incidence is estimated to be 1 in 16 000 live births and 1 in 250 spontaneous abortions. 1
Holoprosencephaly is a malformation sequence with a very variable degree of severity for both the brain and facial abnormalities. Intellectual disability is associated with HPE and seizures are often present.
Case Report
The sonography showed a single ventricular cavity with fusion of the frontal horns of the 2 lateral ventricles, thalami and agenesis of the corpus callosum. (Figure 1)
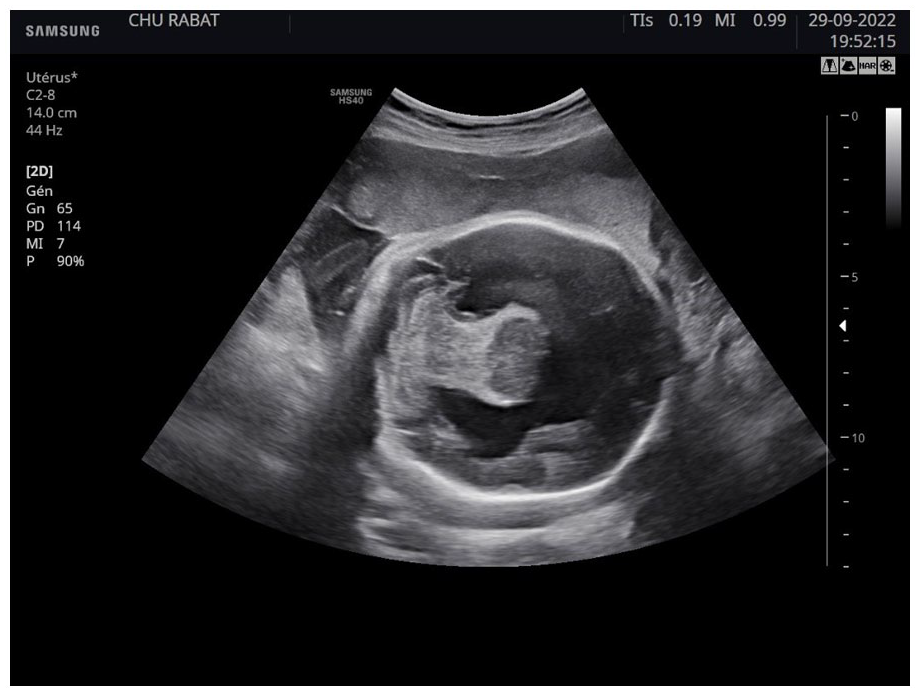
Brain ultrasound showing 2 cerebral hemispheres, with a singular ventricular cavity.

A picture of the newborn with cleft lip and palate.
The brain scan showed: presence of the interhemispheric fissure in the caudal part, with a singular ventricular cavity and hydrocephaly (Figure 3 and Figure 4).

Axial section of brain CT showing the presence of the interhemispheric fissure in the caudal part, with a singular ventricular cavity and hydrocephaly.

Coronal section of brain CT showing the presence of the interhemispheric fissure in the caudal part, with a singular ventricular cavity and hydrocephaly.
Discussion
Classification of holoprosencephaly is based on the complete absence or not of the interhemispheric fissure and the degree of separation of the cerebral hemispheres.
The 4 main types of HPE :
-
-
-
-
The exact cause of HPE is unknown. However, risk factors includes:
✓ Maternal diabetes
✓ Infections during pregnancy like syphilis, rubella, toxoplasmosis, CMV, herpes . . .
✓ Use of certain drugs and medicines during pregnancy
Some children will have an identifiable genetic cause of holoprosencephaly. It can be transmitted in an autosomal dominant way.
Mutation of SHH gene is the most frequent cause of familial holoprosencephaly. 2
About 25% to 50% of HPE are seen in many chromosomal abnormalities, like trisomy 13 and 21. HPE is seen in about 70% of people with trisomy 13, whose birth prevalence is 1/5000. Trisomy 13 can be suspected during pregnancy on ultrasound and confirmed by fetal karyotype test.
Holoprosencephaly is a malformation sequence with a very variable degree of severity for both the brain and facial abnormalities. Children diagnosed with this disorder may have a small head (microcephaly), excessive fluid in the brain (hydrocephalus), facial abnormalities, tooth abnormalities like solitary median maxillary central incisor, cleft lip or palate, epilepsy, and/or endocrine abnormalities. The most severely affected individuals may have cyclopia, a single central eye that is the most severe eye finding seen in holoprosencephaly, though this is very rare. Abnormalities in the formation of the nose may also occur like congenital nasal pyriform aperture stenosis which is a lethal cause of neonatal respiratory distress. 3
Prenatal diagnosis of HPE includes ultrasonography and fetal MRI. The diagnosis could be made in most cases of alobar and semilobar holoprosencephaly after 17 weeks of gestation, when the production of cerebrospinal fluid starts. In lobar cases diagnosis could be difficult because the antenatal picture of septo-optic dysplasia is almost identical to that of lobar holoprosencephaly. 4
The prognosis for individuals with the disorder depends on the severity ad importance of the brain and facial malformations.
There is no standard course of treatment for holoprosencephaly. Treatment is symptomatic and supportive.
Conclusion
This article is a general discussion of holoprosencephaly. We studied 2 cases of semilobar holoprosencephaly: one diagnosed before birth with ultrasonography, and the other after birth with a brain CT.
In conclusion, semilobar holoprosencephaly is a rare structural anomaly of the brain with a complex and multifactorial causes. It needs to be diagnosed prenatally, for its severity and prognosis.
Footnotes
Author Contributions
The author confirms sole responsibility for the following: study conception and design, analysis, and manuscript preparation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
This paper did not involve any research and no ethical clearance was required. A written informed consent was obtained from the patient for the publication of this paper.