
Abstract
The fat mass and obesity-associated gene (FTO) codes for a DNA/RNA demethylase. Pathological variants in this gene are rare, with only three reports in the literature, all with mutations in the catalytic domain. We report the first biallelic human variant in fat mass and obesity-associated gene (c.287G>C, p.Arg96Pro/R96P) outside the catalytic site, causing numerous abnormalities across multiple organ systems, affecting respiratory, cardiovascular, and neurological function. Biochemical assays of cells with the patient’s variant were performed to further quantify the effect of the variant on function. Loss-of-function resulting from the patient’s R96P missense variant was demonstrated with in vitro biochemical characterization of demethylase activity, resulting in a 90% reduction in function of the fat mass and obesity-associated protein compared to wild-type. Our findings demonstrate a novel fat mass and obesity-associated gene non-catalytic site variant with a unique patient phenotype of bilateral multifocal epilepsy and multisystem congenital anomalies.
Keywords
Introduction
Fat mass and obesity-associated gene (FTO) is a Fe2+/2-oxoglutarate-dependent oxidative RNA demethylase, a member of the AlkB family. It has been shown to demethylate 3-meT in single-stranded DNA and 3-meU in single-stranded RNA (ssRNA). It shows greatest activity for N 6 -methyladenosine (m6A, the most abundant methylated base in mRNA) in ssRNA.1–3 The extent of FTO activity on double-stranded DNA differs among literature reports.1,4 Pathogenically viable human variants in FTO appear to be exceedingly rare, with only 11 cases of FTO malformation syndrome currently reported in the literature.5–7
FTO comprises a N-terminal domain (residues 32–326) and C-terminal domain (residues 327–498) with the catalytic site residing in the N-terminal domain. 1 FTO is expressed in most tissues but is highly expressed within the brain and heart. 5 The Human Protein Atlas (proteinatlas.org) highlights FTO RNA expression throughout the brain with protein expression noted particularly in the basal ganglia, cerebral cortex, hippocampus, and cerebellum. 8 Premature cell senescence was also demonstrated in the setting of FTO dysfunction and may be implicated in early lethality. 5
To date, there are 3 published reports of 11 patients total with a “FTO malformation syndrome,” all with reported autosomal recessive inheritance.5–7 Interestingly, these reports identified variants within the catalytic site of the FTO protein at R316Q, S319F, and R322Q. We report the first case of a child with a biallelic c.287G>C (p.R96P) FTO variant and demonstrate the biochemical effects of this variant on FTO enzyme kinetics.
Case presentation
The female infant was born by spontaneous vaginal delivery at 37 weeks 4 days gestation to a G3P3003 mother. Pregnancy was complicated by multiple fetal anomalies in diverse systems as follows: (1) uterine environmental (polyhydramnios, intrauterine growth restriction (estimated fetal weight <1%), umbilical cord thickening with increased volume of Wharton’s jelly, and elevated uterine artery Doppler indices with preserved diastolic flow); (2) central nervous system (CNS; poor gyral development, small cerebral hemispheres, and Dandy-Walker spectrum anomaly with incompletely rotated vermis, large foramen of Magendie, large cisterna magna, and hydrocephalus); (3) musculoskeletal (all long bones <5% mean for gestational age without evidence of fracture, thoracic cage 9% mean for gestational age, and disorganized anterior maxillary tooth buds); and (4) cardiovascular (ventricular septal defect, hypoplastic mitral valve, and aortic arch) (Table 1). Maternal serologies were negative for pathogens associated with congenital malformations. The infant’s mother denied any significant or contributory family history as well as consanguinity.
Phenotypes of patients with reported fat mass and obesity-associated gene mutations.

At birth, the amniotic fluid was meconium stained. The newborn emerged depressed, limp, and apneic, requiring deep suctioning, intermittent positive pressure ventilation, and bag and mask ventilation. Apgars were three at 1 min, 5 at 5 min, and 9 at 10 min. Birth weight was 2.56 kg. The patient was intubated for respiratory failure and pulmonary hypertension. Her physical examination was notable for dysmorphic features, micrognathia, low-set ears, and the fifth toe digit overlapping the fourth digit bilaterally. Genetic analysis showed a normal karyotype and whole genome sequencing identified homozygous c.287G>C (p.Arg96Pro/R96P) variant within the FTO gene. Both parents were heterozygous for this mutation.
Airway evaluation documented severe tracheobronchomalacia. The patient underwent multiple failed extubations, ultimately leading to tracheostomy. She continued to have severe pulmonary hypertension requiring ventilatory support. Postnatal echocardiographic evaluation showed a secundum atrial septal defect, a patent foramen ovale, three pulmonary veins entering the left atrium, and predominant right-to-left flow with reversal of flow through the aortic arch. The patient progressed to severe heart failure due to biventricular systolic dysfunction with an ejection fraction of 18%. However, she was eventually weaned off inotropes.
Focal subclinical seizures (Figure 1) were noted on electroencephalography (EEG) from day 5 of life; the patient’s seizures manifested as episodes of apnea, desaturation, and behavioral arrest. She continued to have multiple daily subclinical breakthrough seizures despite maximum doses of phenobarbital, levetiracetam, and lacosamide along with initiation of a ketogenic diet (maximum beta-hydroxybutyrate achieved 2.04 mmol/L).

Seizure seen on day 5 of life. (A) EEG shows rhythmic sharp-wave activity over the midline, left and right parasagittal regions, spreading to the left hemisphere, 4 s later to the right hemisphere. Sensitivity at 15 µV/mm. Timebase 15 mm/s. B. Seizure seen at the age of 3 months. EEG shows high voltage rhythmic delta evolving into sharp waves over the right temporal region. Sensitivity at 10 µV/mm. Timebase 30 mm/s. C. Seizure at the age of 7 months. EEG shows high voltage rhythmic spike-wave activity over the right hemisphere, maximal over the right temporal. Independent spikes seen over the left temporal. Sensitivity at 20 µV/mm. Timebase 30 mm/s. D. Seizure at the age of 7 months. EEG shows high voltage rhythmical delta activity with onset over the left temporal region. Sensitivity at 20 µV/mm. Timebase 30 mm/s. E. Interictal EEG on day 5 of life shows awake background, continuous with multifocal delta slowing and sharp waves. Sensitivity at 7 µV/mm. Timebase 15 mm/s. F. Interictal EEG at the age of 3 months shows continuous background and multifocal polymorphic delta slowing, mainly over the bilateral posterior quadrants. Sensitivity at 10 µV/mm. Timebase 30 mm/s.
The patient’s magnetic resonance imaging (MRI) brain was notable for Dandy-Walker malformation, hydrocephalus, a thin cortical mantle, and severe loss of the underlying parenchyma (Figure 2). Third ventriculostomy and choroid plexus cauterization were undertaken for management of the hydrocephalus.
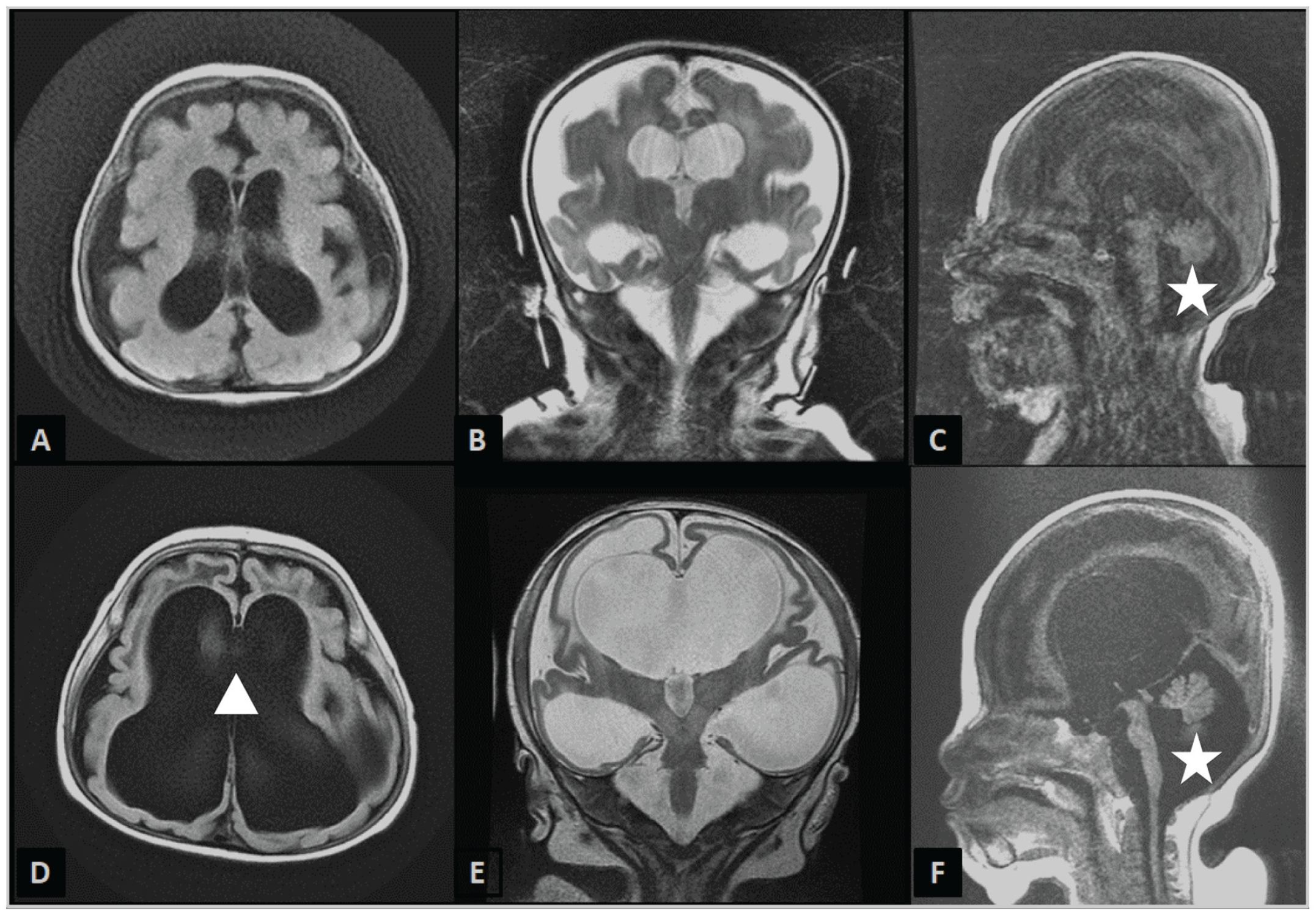
Representative brain MRI findings at 2 months (A–C) and 8 months (D–F) demonstrating the Dandy-Walker malformation (white star) (C, F) and the severe progressive ventriculomegaly (white triangle). There is marked symmetrical diminution and gliosis of the cerebral white matter. A, D: Axial T2 FLAIR (fluid-attenuated inversion recovery) images. B, E: Coronal T2-weighted images. C, F: Sagittal T1-weighted image.
The patient has since been discharged home tracheostomy and is ventilator dependent. At 14-month follow-up, she was being treated with vigabatrin 500 mg q12, brivaracetam 30 mg q12, levetiracetam 400 mg q12, and phenobarbital 22.4 mg q12 via gastrostomy tube. No clinical seizures have been noted for 4 months; however, intermittent respiratory distress is noted requiring frequent suctioning. As of writing, the patient is 16 months old, having made minimal developmental progress, with examinations documenting ongoing global developmental delay and ventilator dependence.
To further evaluate the effect of this patient’s novel FTO variant on enzymatic function, biochemical kinetics assays were performed with R96P variant FTO protein. 200 ng purified polyadenylated RNA from HEK293T cells was incubated with 2 µM FTO (WT, R96P, and catalytically dead mutant R316Q/R322Q, denoted as dQ) for 1 h in 20 µl solution under the reported demethylation conditions. 9 For the demethylation-time curve, FTO was incubated with ssRNA probe with a GGm6ACU sequence. The reaction was quenched by iron chelation with ethylenediaminetetraacetic acid which inactivates FTO, and RNA was isolated by Trizol reagent and subjected to liquid chromatography–mass spectrometry (LC-MS/MS) measurement. Analysis was performed using a Student’s t-test.
Biochemical results
Biochemical kinetics assays revealed that R96P mutant FTO resulted in almost complete loss of m6A conversion relative to wild type and comparable to known catalytically dead mutant FTO proteins (dQ) (Figure 3). Over a 120-min timescale, no greater than 10% conversion of m6A was achieved with the R96P mutant relative to complete conversion with wild-type FTO. These results suggest that the patient’s novel variant results in severely impaired FTO function.

In vitro FTO-catalyzed demethylation is markedly decreased in R96P mutant protein. A. Quantification of methylation percentage of the m6A/A ratio by LC-MS/MS in vitro. Error bars represent mean ± s.d., n = biological replicates. *p < 0.05. ****p < 0.0001. n.s. represents not significant. B. FTO-catalyzed demethylation (%, wild type (WT) and R96P) versus time (min) toward m6A in GGm6AU-containing synthetic ssRNA probes. Error bars represent mean ± s.d., n = 3 biological replicates.
Discussion
To date, there are three reports implicating variants in FTO as a cause of human pathology. The phenotype of published reports and that of our patient are compared in Table 1. Boissel et al. reported a severe phenotype in nine individuals within one consanguineous Palestinian Arab family as a result of c.947G>A (p.R316Q) variant. 5
Daoud et al. reported a 21-month-old female infant with a homozygous c.956C>T (p.S319F) variant. 6 The patient’s MRI showed decreased brain parenchyma, delayed myelination, and a thin corpus callosum.
Rohena et al. reported two siblings from a consanguineous Yemini family with a homozygous c.965G>A (p.R322Q) pathogenic variant. 7 The first child had a normal prenatal course but was small for gestational age. She remained in the neonatal intensive care unit (NICU) for 2 months postpartum due to hypotonia, respiratory failure, and poor feeding. Brain MRI confirmed ventriculomegaly with additional findings of bilateral periventricular hyperintensities consistent with zones of demyelination. Repeat imaging at age 4 months showed cortical atrophy. The child died of respiratory failure at the age of 6 years due to recurrent pneumonias.
In the second child, fetal ultrasound demonstrated an abnormal aortic arch, subsequently shown to be a left-sided cervical aortic arch with aneurysmal segments. Birth weight was appropriate. She spent 1 month in the NICU for poor feeding and required nasogastric feeds on discharge. Her 1-year follow-up disclosed multiple abnormal physical features (see Table 1), and truncal hypotonia with mild spasticity of upper and lower extremities. She died of respiratory failure at the age of 2.5 years.
Our patient showed several similarities to those previously described in the literature (see Table 1). All cases appeared to have a severe CNS phenotype. Ventriculomegaly and cortical atrophy were common to both this patient and prior cases. Seizures were noted in several cases (although unfortunately type and semiology were not noted in past reports), but none proved as resistant to medical control of the seizures as was our patient, whose seizures remained uncontrolled and frequent despite three antiepileptic drugs at maximal doses and a ketogenic diet; in this case, the resistant seizures may have been in part due to the patient’s significant brain malformations. Cardiac abnormalities and respiratory failure were common to most patients and were also seen in this case. All patients described died at a young age.
Unlike the previously reported cases, this patient’s variant is related to an area of the protein predicted to be the “substrate recognition lid,” a conserved region that is responsible for substrate fixation and selectivity remote from the catalytic site. 4 This variant was found in two participants within the 2866 individuals sequenced by Meyre et al. 10 R96M (methionine) and R96Q (glycine) mutants were shown to greatly compromise the demethylase activity of FTO in vitro. 4 Our case helps to expand the scope of the literature surrounding this rare disorder and adds to the clinical phenotypes that comprise the FTO malformation syndrome with a specific focus on intractable epilepsy.
There are several possible explanations for the development of the structural abnormalities seen in FTO malformation syndrome. It can be argued that the aberrant neural development seen in these cases may also be due to the many cardiac and respiratory anomalies also present in this syndrome. While this is possible, Gao et al. have demonstrated that in knockout mice with a deletion of neural FTO only, a similar phenotype of neural growth retardation is observed, 11 suggesting that FTO is key for the proper development of the CNS.
The profound structural malformations seen in these patients may also explain the marked epileptic activity that we have described, but notably, FTO has previously been implicated in epileptogenesis. 12
Conclusion
In conclusion, we present a novel genotype–phenotype addition to the FTO malformation syndrome in human newborns and demonstrate biochemically proven loss-of-function due to an extra-catalytic FTO variant. Further work is required to demonstrate the genome-wide and tissue-specific effects of this likely pathogenic variant.
Supplemental Material
sj-docx-1-sco-10.1177_2050313X231188883 – Supplemental material for Case report: A novel biallelic FTO variant causing multisystem anomalies with severe epilepsy, widening the spectrum of FTO syndrome
Supplemental material, sj-docx-1-sco-10.1177_2050313X231188883 for Case report: A novel biallelic FTO variant causing multisystem anomalies with severe epilepsy, widening the spectrum of FTO syndrome by Naomi Mayman, Jiangbo Wei, Shangjun Cai, Rohan Soman, Hillary Raynes, Maite La Vega-Talbott, Chuan He, Thomas Naidich, G. Praveen Raju and Sathiji Kathiresu Nageshwaran in SAGE Open Medical Case Reports
Footnotes
Acknowledgements
The authors would like to thank the patient’s family for graciously allowing the publication of their child’s case.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent statement
Written informed consent was obtained from a legally authorized representative for anonymized patient information to be published in this article. Institutional Review Board of the Mount Sinai School of Medicine, in accordance with Mount Sinai’s Federal Wide Assurances (FWA#00005656, FWA#00005651) to the Department of Health and Human Services approved the following human subject research. HS#:STUDY-21-01960.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.