
Abstract
Over the past two decades, significant technological advances have facilitated the identification of hundreds of genes associated with hearing loss. Variants in many of these genes result in severe congenital hearing loss with profound implications for the affected individual and their family. This review collates these advances, summarizing the current state of genomic knowledge in childhood hearing loss. We consider how current and emerging genetic technologies have the potential to alter our approach to the management and diagnosis of hearing loss. We review approaches being taken to ensure that these discoveries are used in clinical practice to detect genetic hearing loss as soon as possible to reduce unnecessary investigations, provide information about reproductive risks, and facilitate regular follow-up and early treatment. We also highlight how rapid sequencing technology has the potential to identify children susceptible to antibiotic-induced hearing loss and how this adverse reaction can be avoided.
Introduction
In 1997, a team of researchers identified a large family from the Mirpur region of Pakistan in which multiple members were affected by nonsyndromic hearing loss (NSHL; Kelsell et al., 1997). Using linkage analysis and Sanger sequencing, the authors identified a common pathogenic variant in GJB2, a gene, which encodes a protein called connexin-26, that was responsible for the hearing loss in this family. At the same time, major sequencing facilities around the world were collaborating to methodically analyze the whole sequence of the human genome. The Human Genome Project to sequence a complete human genome had been formally underway for 7 years but was not finished until 2003, at a total cost of approximately $3 billion (Green, Watson, & Collins, 2015). Just over 20 years since the discovery of GJB2 as a cause of NSHL and 15 years since completion of the Human Genome Project, the landscape of hearing loss genetics has changed fundamentally. To date, over 100 genes have been associated with NSHL (https://hereditaryhearingloss.org), and the capacity to identify pathogenic variants in those genes has increased exponentially. Both the speed of variant identification and the cost of DNA sequencing have improved dramatically; genetic variants associated with hearing loss can now be identified at the bedside in minutes and sequencing a whole genome now costs less than $1,000.
Hearing loss affects between 1 and 2 newborns per 1,000 and can have a significant impact on a child’s communication, quality of life, and educational attainment (Roland et al., 2016; Teasdale & Sorensen, 2007; Wood, Sutton, & Davis, 2015). As such, prompt recognition and accurate diagnosis are crucial to optimize outcomes. The etiology of hearing loss is manifold but can be typically divided into either environmental or genetic causes. The most prescient environmental exposures include neonatal or prenatal infections, extreme prematurity, and exposure to ototoxic agents, either during pregnancy or in the neonatal period (C. C. Morton & Nance, 2006; Robertson, Howarth, Bork, & Dinu, 2009). Meanwhile, approximately 50% of all cases of neonatal deafness are caused by an underlying genetic variant (Smith, Bale, & White, 2005). Children might carry a genetic variant resulting in NSHL or syndromic hearing loss (SHL), or they might harbor a genetic variant that predisposes to drug-induced ototoxicity.
In cases of SHL or NSHL, early identification and confirmation of the genetic variant facilitates patient management. If a patient is found to harbor a variant in a gene associated with NSHL, the family can be counseled accordingly, and management can focus on the auditory system. If the gene, or even the specific variant, is associated with syndromic disease, appropriate surveillance can be initiated to monitor the development of additional nonhearing-associated complications. This appreciation of genotype–phenotype correlation is increasingly allowing clinicians to develop personalized management plans and more relevant genetic counseling for patients and families (Parker & Lucassen, 2004).
Advances in sequencing capabilities and understanding of the genome have begun to radically alter our ability to identify and interpret genetic variants, leading to more rapid diagnosis, improved patient monitoring, and, in some cases, avoidance of hearing loss. Here, we highlight the increasing relevance of genetics in the diagnosis, management, and prevention of hearing loss in the neonatal period.
Role of Genetic Testing in the Diagnosis of Hereditary Hearing Impairment
Estimates have previously suggested that at least 1% of all human genes encode a protein relating to hearing function with varied roles pertaining to the morphogenesis or maintenance of structures in the auditory pathway (Friedman & Griffith, 2003). Hearing loss can be broadly classified as nonsyndromic, without extra-auricular features, or SHL, where the phenotype can involve a diverse range of body systems external to the auditory pathway (Smith et al., 2005). NSHL is the most common clinical presentation, accounting for 70% of congenital cases (C. C. Morton & Nance, 2006).
Variants in hearing loss-associated genes can be inherited in an autosomal-recessive, autosomal-dominant, X-linked, or a mitochondrial pattern (Smith et al., 2005). The affected gene, or even the specific variant, often correlates with a distinct clinical phenotype including age of onset, phenotypic severity, disease progression, and the auditory frequencies affected (Snoeckx et al., 2005; Taylor et al., 2013). The majority of prelingual onset hearing loss follows an autosomal-recessive inheritance, affecting early speech development, whereas autosomal-dominant forms are more frequently associated with postlingual deafness (Sloan-Heggen et al., 2016). Accurate clinical phenotyping and a thorough family history can therefore be useful in establishing and refining a list of candidate genes. However, the relationship between genotype and phenotype is not always consistent and hearing loss disorders demonstrate significant phenotypic overlap (Hoefsloot, Feenstra, Kunst, & Kremer, 2014). In addition, locus heterogeneity is a feature of certain syndromes, meaning that some disorders can result from variants in several genes, such as in Usher syndrome (MIM 276900; Keats & Savas, 2004). Furthermore, variants in certain genes, for example, GJB2, are associated with both dominantly and recessively inherited hearing loss (Snoeckx et al., 2005). Because of this phenotypic and genetic heterogeneity, clinical features and family history are rarely sufficient to predict the causative gene. This has led to the development of gene panels as part of a wider testing strategy, where a number of genes or genetic variants can be assessed in a single assay.
Targeted Gene Panels
The British Association of Audiological Physicians (BAAP, 2015) has produced guidelines for the etiological assessment of bilateral sensorineural hearing loss in children (Figure 1). Level 1 investigations include the clinical history and examination, audiograms of the family, ophthalmological assessment, cytomegalovirus (CMV) testing, an electrocardiogram, brain imaging, urine analysis, and testing for variants in GJB2. This initial screen is designed to identify the most common causes of bilateral hearing loss in childhood. The electrocardiogram is used to screen for the presence of a prolonged QT interval, indicative of Jervell and Lange-Nielsen syndrome (MIM 220400), whereas proteinuria might suggest a diagnosis of Alport’s syndrome (MIM 301050). Ophthalmological assessment can reveal a number of abnormalities, which can indicate syndromic diseases such as Usher (MIM 276900), CHARGE (MIM 214800), or Waardenburg (MIM 193500) syndromes. Although these extra-auricular features can be extremely useful in helping to establish a differential diagnosis, their absence has the potential to be misleading in determining a precise diagnosis. The characteristic coloboma of CHARGE syndrome is absent in approximately 20% of patients, while the proteinuria in Alport’s syndrome is progressive, meaning this feature will often be absent in younger patients (Trider, Arra-Robar, van Ravenswaaij-Arts, & Blake, 2017).
The British Association of Audiological Physicians has produced guidelines for the etiological assessment of bilateral sensorineural hearing loss in children. The left side of the schematic demonstrates the current stepwise testing strategy of a child with hearing loss via Level 1 and Level 2 tests in this guideline. The right side of the schematic displays how emerging genetic technologies might be implemented in future testing strategies. Note. ECG = electrocardiogram; WES = whole exome sequencing; POCT = Point of care Test; AIHL = Antibiotic Induced Hearing Loss; USS = Ultrasound Scan; WGS = Whole Genome Sequencing.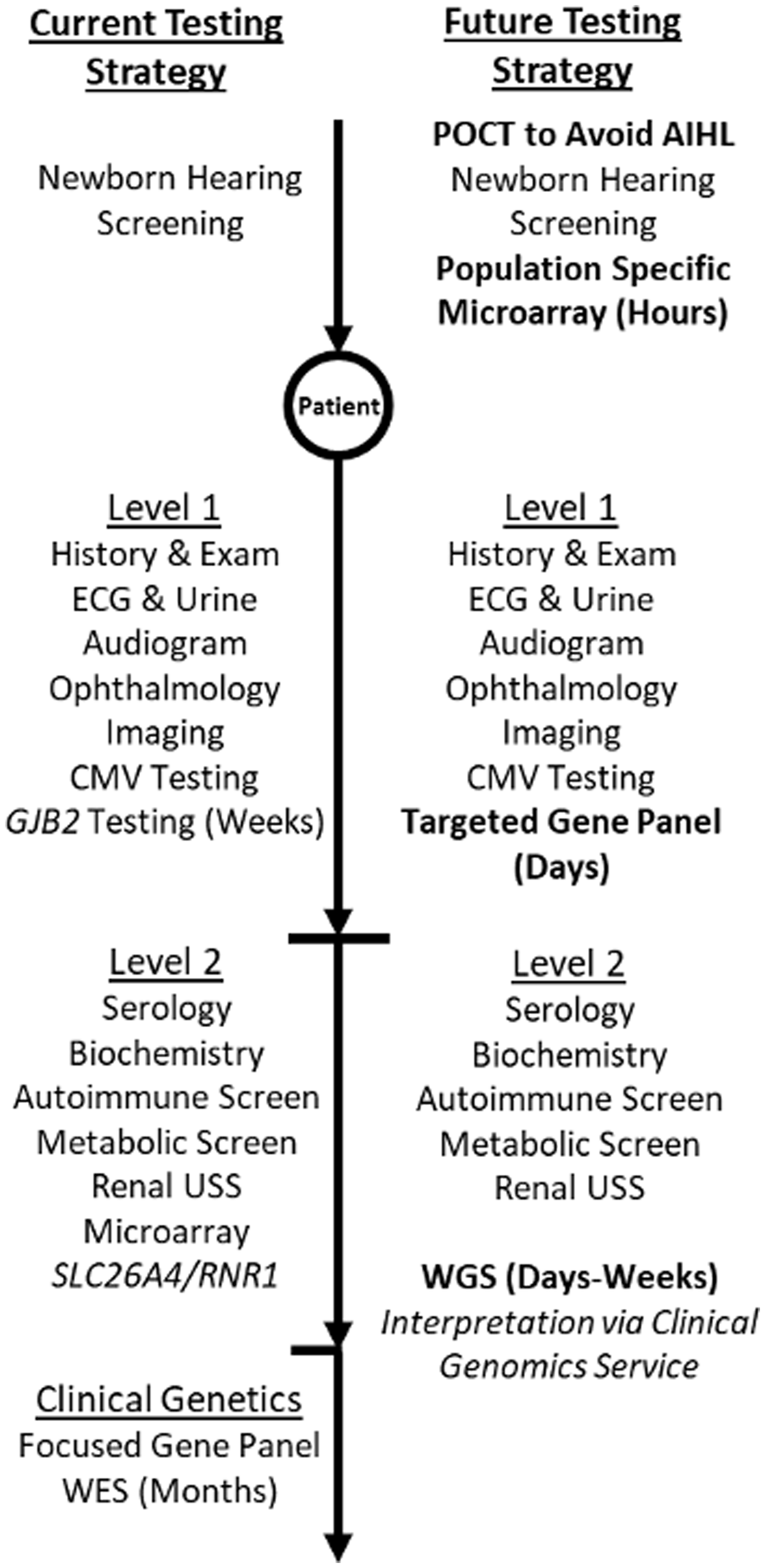
Due to the prevalence of GJB2 variants in NSHL, GJB2 testing is included in Level 1 investigations, but no other genes are assessed at this level. If no etiological cause is identified from the Level 1 testing, progression to Level 2 testing includes consideration of microarray comparative genomic hybridization to identify chromosomal alterations, including deletions and duplications and single gene testing for variants in SLC26A4 (MIM 605646), EYA1 (MIM 601653), and OTOF (MIM 603681), the respective causes of Pendred (MIM 274600) and branchiootorenal (MIM 113650) syndromes and autosomal-recessive auditory neuropathy (MIM 601071). If the child or the mother has been exposed to aminoglycoside antibiotics, then testing for the m.1555A>G variant is appropriate as this variant is associated with aminoglycoside-related ototoxicity. If these further investigations fail to reveal the cause of the hearing loss, further genetic testing, often using a gene panel-based approach, can be performed.
Most modern gene panels utilize next-generation sequencing technology, a catch-all term for a range of approaches that allow for massively parallel high-throughput sequencing, providing results significantly quicker than traditional methods (Table 1). Targeted panels typically constrain their sequencing to a predefined set of genes, decided upon by the operator. There are a number of commercially available panels, which vary in their methodology and in the number of genes targeted (Sloan-Heggen & Smith, 2016). Since 2001, the UK Genetic Testing Network (n.d.) has published the NHS Directory for Diagnostic Testing, which evaluates and recommends genetic tests commissioned in the NHS. At present in the United Kingdom, an inherited hearing loss genetic panel has been approved to analyze the coding regions and intron/exon boundaries of 95 genes. Confirmatory testing of any variant identified is then performed via Sanger sequencing to facilitate testing in other family members.
A Comparison of Different Genetic Testing Strategies.

Whole Genome Sequencing
The BAAP guidelines in 2015 acknowledged that widespread genetic testing for deafness is likely to become available with the implementation of next-generation sequencing, where large numbers of genes can be sequenced rapidly and cost-effectively. As such, guidelines for further genetic testing are likely to evolve in coming years and genetic testing will become increasingly accessible earlier in the diagnostic process. In 2018, the NHS National Genomics Service was launched creating a genomic laboratory service with a national genomic test directory to promote accessible, equitable, and cost-effective genetic testing across England. When fully operational, this will facilitate comprehensive access to genetic testing for individuals, especially children, with NSHL.
WGS encompasses both coding and noncoding regions, including the other 98% of the genome not captured by WES (Table 1). These noncoding (intronic) regions were historically considered junk DNA, with little relevance other than as a place holder for the protein coding exons (Franchini & Pollard, 2017). It is now understood that these intronic regions have vital roles and that noncoding variants can lead to disease (Check, 2006). In a consanguineous family from Saudi Arabia with a clinical diagnosis of Usher syndrome, standard genetic testing failed to reveal a causative variant in any of the associated genes. WGS meanwhile identified a deep intronic variant, which created a cryptic splice site causing loss of protein resulting in the phenotype (Khan et al., 2017). Cases such as this highlight the relevance of WGS and its additional diagnostic value beyond WES or panels. Efforts to introduce WGS into clinical practice have been supported by initiatives such as the 100,000 Genomes Project in the United Kingdom and the SEQuencing a Baby for an Optimal Outcome trial in the United States (C. Morton, 2015). This project is currently underway at the Brigham and Women’s Hospital and aims to establish the efficacy of performing WGS in all babies who fail their newborn screening assessment. Considering the falling cost of sequencing alongside the current enthusiasm for this approach, it is foreseeable that WGS will become standard of care for individuals with severe to profound hearing loss after negative Level 1 and Level 2 investigations (Figure 1).
Rapid Genetic Sequencing
The utilization and impact of genetic information, by the very nature of sequencing technology, have always been in the context of diagnosing chronic diseases such as rare syndromic disorders and cancers. Time between sample collection and delivery of a clinically relevant result is often measured in months rather than minutes. The concept that genetic information might be generated in real time and influence treatment decisions, especially in the acute setting, has been infeasible. However, novel sequencing approaches have been developed in recent years and have the potential to provide clinically actionable results in a clinically relevant timeframe.
Microarray Chip-Based Sequencing
In parallel to advances in WGS and WES, chip-based microarray technology has become increasingly available. These generate data that are often straightforward to interpret, rapid to undertake, and are cost-effective. Although there are over 100 disease-causing genes associated with congenital hearing loss, specific variants within certain genes account for a larger proportion of cases. The GJB2 c.35delG variant is common across Western Europe causing an estimated 75% of all GJB2-related NSHL, while the SLC26A4 p.Leu236Pro (c.707T>C) and the p.Thr416Pro (c.1246A>C) variants are also widely prevalent across the same regions, causing 23.1% and 12.8% of all SLC26A4-related deafness in the United Kingdom (Tsukada, Nishio, Hattori, & Usami, 2015). Targeted assays can now be developed to cheaply and rapidly identify specific variants, common within a population. The CapitalBio-Miami-Oto-Array (CapitalBio Corporation, Beijing, China) has been designed, using microfluidic technology, to identify nine of the most common variants in patients of European descent within four disease-causing genes, GJB2, GJB6, SLC26A4, and the mitochondrial gene, RNR1 (Yan et al., 2017). This technology facilitates an initial screen of the most common genetic causes of NSHL for less than $30 in approximately 4 hr.
Targeted variant genotyping could well form part of an updated BAAP guideline as a new Level 1 investigation. Alternatively, this approach might be considered as part of newborn screening program as a supplement to the newborn hearing test, where genotyping would be undertaken on blood spots from all babies. Potentially, this would identify newborns that pass the newborn hearing screen but are at an increased risk of childhood-onset NSHL and so should undergo more regular assessment and earlier intervention (Figure 1). A formal health economic evaluation and determination of acceptability and feasibility would be required for implementation of this technology but given low cost and rapid turnaround, this requires serious consideration.
It is important to consider ethnic variation when designing targeted variant panels to be used more widely for the investigation of hearing loss. Although the GJB2 c.35delG variant is common across Western Europe, its prevalence is negligible across India, China, and across Southeast Asia. Instead, these ethnic groups have different common genetic variants associated with hearing loss (Tsukada et al., 2015). The vast majority of genomes sequenced to date are from individuals of White European descent. This means that the genetic knowledge of certain disorders may be less mature or even not relevant in other populations. There are now international efforts to diversify genomic datasets by sequencing genomes from greater numbers of individuals without European ancestry. When designing chip-based assays for hearing loss, it is important to consider population diversity and several different population-specific chips may be required to ensure appropriate coverage.
Point of Care Testing
Many commonly prescribed medications have the potential to cause ototoxicity, particularly if dispensed over a protracted period or in excess. Aminoglycosides are broad-spectrum antibiotics, which act by binding to the 16S rRNA component of the bacterial 30S ribosomal subunit, resulting in the translation of truncated proteins (Lanvers-Kaminsky & Ciarimboli, 2017; Schacht, Talaska, & Rybak, 2012). These abnormal proteins stimulate a stress response within the bacteria culminating in cell death. Due to their low cost and effectiveness, they are one of the most frequently prescribed medicines globally, used in cases of Gram-negative sepsis, pseudomonas colonization, and treatment of multidrug-resistant tuberculosis (Bitner-Glindzicz & Rahman, 2007). The National Institute for Health and Care Excellence (NICE) advises the use of intravenous benzylpenicillin with gentamicin as the first-choice antibiotic regimen for empirical treatment of suspected infection in the neonatal period (NICE CG149). This combination has the major advantage of having a narrow spectrum of activity and lower risk of antibiotic resistance compared to alternative antibiotic regimens.
The side effect profile of aminoglycoside antibiotics is well known, with nephrotoxicity, vestibulotoxicity, and ototoxicity commonly recognized in cohorts who receive large amounts of aminoglycoside (Prayle & Smyth, 2010). Careful analysis revealed that aminoglycoside-induced ototoxicity clusters within families and this trait was transmitted maternally, consistent with mitochondrial inheritance. Further work demonstrated that this susceptibility was caused by a variant in the 12S rRNA (RNR1) m.1555A>G (Prezant et al., 1993). This variant causes a change in the conformation of the 12S rRNA, producing a structure more similar to the bacterial 16S rRNA.
The m.1555A>G variant is strongly associated with aminoglycoside-induced ototoxicity and has a reported prevalence of 0.2% (∼1 in 500). As part of a wider study, we accessed data from 119,845 individuals within the U.K. Biobank dataset, establishing a U.K. wide prevalence of 0.198% (95% confidence interval: 0.173%–0.225%) for the variant. It has previously been suggested that genetic testing should be used in children requiring aminoglycosides to prevent hearing loss and that this approach would be cost-effective when balanced against the costs of lifelong deafness (Bitner-Glindzicz et al., 2009; Bitner-Glindzicz & Rahman, 2007). Currently, in many centers, patients with cystic fibrosis are tested for the variant preemptively as it is anticipated that these individuals will require aminoglycoside antibiotics to treat Gram-negative respiratory infections at some stage during their illness. This is currently undertaken using pyrosequencing, which takes up to 7 days to return a result from the laboratory. Understandably, this approach is not suitable for use in neonates with suspected sepsis where a prescribing decision is required within an hour.
At present, there is an inability to test for the m.1555A>G variant in the acute setting such as babies admitted to the neonatal intensive care unit, representing approximately 90,000 admissions per year in the United Kingdom alone. Based on the prevalence of the variant, approximately 180 cases of deafness could be avoided per year, if clinicians were able to identify carriers and prescribe an alternatively efficacious antibiotic regimen. We have developed a loop-mediated isothermal amplification assay for buccal swabs on a point-of-need rapid thermocycler platform, which can identify the m.1555A>G variant in ∼22 min (Duffy et al., 2017).
The Pharmacogenetics to Avoid Loss of Hearing trial has been designed to determine the feasibility of implementing this assay in the acute setting. This represents the first study where a genetic test is being used to impact patient management in this context. Over a 6-month period, every admission to two large neonatal intensive care units in Manchester and Liverpool will receive this genetic test on admission to the neonatal intensive care unit and antibiotic prescribing will be tailored depending on the presence or absence of the variant.
Studies such as this serve to demonstrate a dramatic shift in our ability to leverage the power of genetics in everyday healthcare. By utilizing a novel genetic technology, we can now deliver a clinically relevant genetic result within a clinically relevant timeframe, avoiding a major cause of neonatal ototoxicity in the acute setting.
Conclusions
Over the past two decades, marked advancements in genetic technologies have facilitated our appreciation of the rich genetic variation underpinning childhood hearing loss. Next, we must ensure the equitable and comprehensive introduction of clinical genomic testing allowing early diagnosis and facilitating optimum clinical care. In some cases, such as by avoiding adverse drug reactions, adoption of rapid genetic screening may prevent certain forms of hearing loss completely. The approaches will develop as technologies improve, costs alter, and different treatment options become available.
Footnotes
Author's Note
Iain A Bruce is also affiliated with Division of Infection, Immunity and Respiratory Medicine, School of Biological Sciences, University of Manchester, UK and Royal Manchester Children's Hospital, Manchester University NHS Foundation Trust, UK.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Hearing loss work in the Newman lab is funded by Action Medical Research (GN2494) and NIHR i4i (II-LB-0417-20002) and Manchester NIHR Biomedical Research Centre (IS-BRC-1215-20007). Development of the Pharmacogenetics to Avoid Loss of Hearing project was funded by Action on Hearing Loss (F67_Newm). Leslie P Molina-Ramírez is sponsored by the National Council of Science and Technology (Consejo Nacional de Ciencia y Tecnología CONACyT - Mexico City). CCM acknowledges support by the Manchester NIHR Biomedical Research Centre and NIH/NIDCD grant DC015052.