
Abstract
Progressive myoclonus epilepsies (PMEs) are a group of genetic disorders marked by myoclonus, epilepsy, and progressive neurological decline. Unverricht–Lundborg disease (ULD) is among the more common forms, though prevalence varies. We present the case of an 8-year-old male with infantile-onset ULD, notable for significant developmental delay and recurrent seizures. The patient developed early myoclonic jerks and tonic seizures, accompanied by cognitive impairment and global developmental delay. Family history revealed similar neurological challenges in siblings. Genetic testing confirmed a homozygous missense pathogenic variant in the cystatin B (CSTB) gene. With antiepileptic medications, seizure control improved, highlighting the role of early therapeutic intervention. ULD results from loss-of-function mutations in CSTB, leading to CSTB deficiency and impaired protease regulation. Although progressive in nature, management with appropriate therapy can reduce seizure burden and improve quality of life. This case emphasizes that PME may present early in childhood with tonic seizures and cognitive dysfunction, and early recognition is essential for optimal care.
Keywords
Introduction
Progressive myoclonus epilepsies (PMEs)—a set of genetic diseases with 3 main clinical features: myoclonus, epilepsy, and progressive neurological decline, particularly dementia and ataxia. Despite sharing the same name, PMEs have different prognoses, etiologies, and pathogenesis.1-3 The most prevalent forms of PME include neuronal ceroid-lipofuscinoses, sialidosis, Lafora’s disease, myoclonus epilepsy with ragged red fibers (MERRF), and Unverricht–Lundborg disease (ULD), which is the purest, least severe, and not associated with progressive cognitive deficit. The most common degenerative forms are ULD and MERRF. “Degenerative” refers to types in which there is no indication of neuronal storage and only neuronal loss and gliosis in light microscopy of the brain. 4
Cystatin B (CSTB) plays an endogenous neuroprotective role in ULD, a neurodegenerative disease. The susceptibility of different neuronal populations to CSTB deficiency varies. Most patients have the expansion mutation in homozygous form, particularly in populations where there is a founder effect. The remaining patients experience compound heterozygosity, where a more uncommon mutation alters the transcript’s structure. 5
ULD, also called PME type 1 (EPM1), is usually characterized by the presence of ataxia associated with myoclonus and epileptic seizures without progressive cognitive deficit, presenting during late childhood and early adolescence between 8 and 15 years of age. The condition is progressive at first; however, with current anti-seizures medications, myoclonus may eventually settle or even improve. Typically, the symptoms regress as individuals get older. Patients may experience a cessation of tonic seizures and develop coping mechanisms for myoclonic jerks. The prognosis for ULD patients has greatly improved due to advances in medicine and adjustments to the overall therapeutic regimen. By demonstrating that the CSTB gene, which underlies ULD, harbors a disease-causing mutation, the clinical diagnosis of UDL can now be confirmed. 6
While ULD has been described in prior reports, this case is distinct due to its early onset, more severe, and dominant presentation. It represents an 8 years old child whose early-onset phenotype was identified at 3 months of age particularly involving marked myoclonic jerks followed by tonic seizures, as well upon further assessment, the patient exhibits significant delay in developmental milestones and cognitive impairment with no evidence of symptoms improvement nor regression. A homozygous mutation G4R in the CSTB gene was found in the exome of the patient and his deceased siblings who all had passed away at a young age through genetic analysis. Our case report highlights the intra-familial age and symptoms diversity and may help to expand the age and clinical boundaries for this disease. Even in patients who are very young, the diagnosis of ULD should be taken into consideration if there is a suggestive family history. No comparable cases have been reported.
This case has been reported in line with CARE criteria. 7
Case Presentation
An 8-year-old male patient presented with a history of seizures. The patient’s medical history began at the age of 3 months, marked by the onset of myoclonic jerks followed by tonic seizures occurring weekly for a month, each lasting between 30 and 60 seconds. Both the patient’s and the family’s medical histories were evaluated, and this information led to the adoption of specific management strategies aimed at controlling seizure activity and preventing further occurrences.
When the patient presented to our hospital at the age of 7, he exhibited significant developmental delay and cognitive impairment. Physical examination revealed axial hypotonia, spasticity in the extremities with contractures in the elbows and knees, limited hip abduction and knee extension, increased deep tendon reflexes, and a positive Babinski sign. He also had profound motor impairments, including long-standing inability to ambulate and severe upper extremity dysfunction. Additionally, notable cognitive deficits are observed, manifesting as impaired attention, markedly reduced eye contact and severe expressive aphasia, indicating significant challenges in using and comprehending language.
The patient has 3 siblings, 2 sisters, and 1 brother, all of whom had ULD. The deceased brother, who passed away at 10 years old, exhibited delayed developmental milestones and began experiencing seizures at the age of 8 months. His symptoms included generalized hypotonia and dystonic posturing, along with excessive hand and leg movements, limited eye contact, and microcephaly. Similarly, the deceased older sister, who died at 5 years old, also had delayed developmental milestones and seizures. Additionally, she demonstrated poor responses to sounds, teeth grinding, and microcephaly while exhibiting autistic behaviors like stereotypical hand movements and rocking. Meanwhile, the surviving sister, currently 12 years old, presents with spastic limbs, increased deep tendon reflexes, and dystonic upper limb movements. She struggles with eye contact, tracking moving objects, and grasping, along with challenges in social interaction and left-sided esotropia. No other family members are affected.
Due to the familial history of similar presentations among siblings, suspicions of an autosomal recessive genetic disorder arose. Subsequent whole exome sequencing confirmed a homozygous missense pathogenic variant in the CSTB gene, thereby solidifying the diagnosis of the Unverricht–Lundborg type of PME (Figure 1).
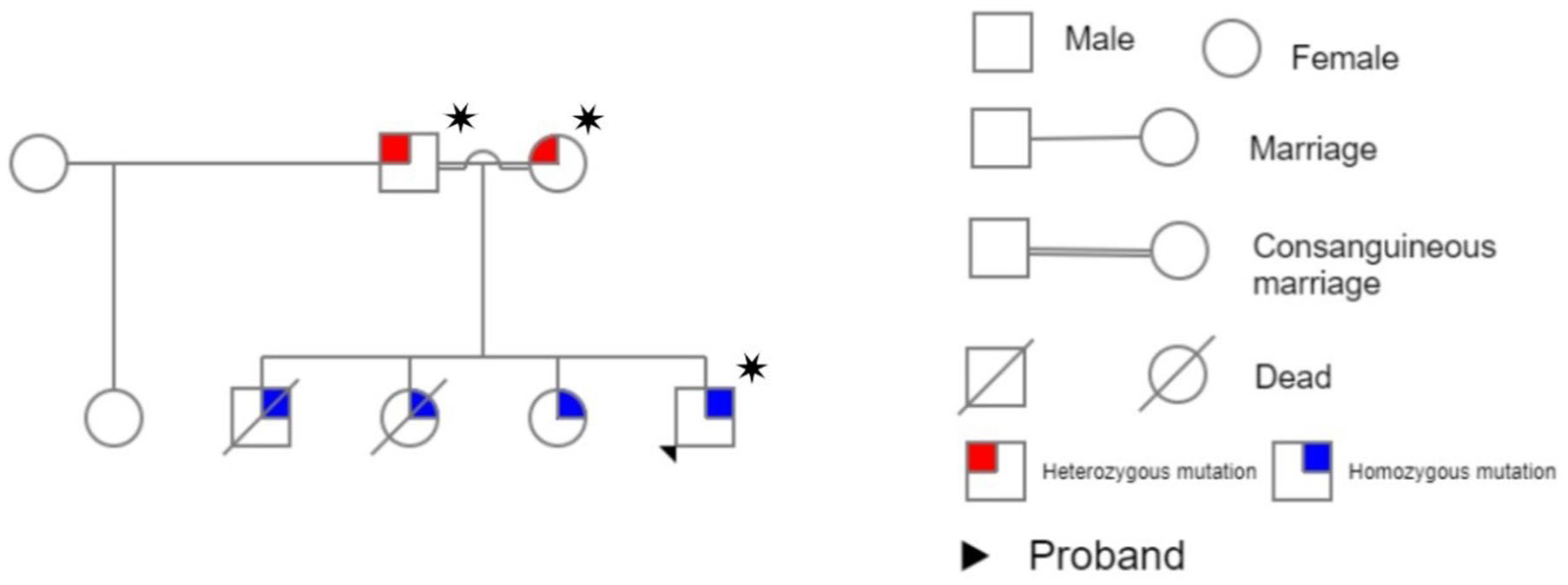
Pedigree of the family. Patients who were tested for the mutation are marked with a star sign.
Following the diagnosis, the patient was treated with sodium valproate (1000 mg/day), resulting in a notable improvement in seizure activity. The patient also received physiotherapy, which improved his spasticity.
Discussion
EPM1, also referred to as ULD, is a rare autosomal recessive neurodegenerative disorder characterized by myoclonus, epilepsy, and ataxia, while cognitive function is generally preserved. 6 The disease typically manifests in late childhood or adolescence, with its onset ranging from 6 to 18 years of age, which contrasts with our case, which presented at 3 months of age. ULD has the highest incidence worldwide among patients with PME. It is relatively frequent in Western Mediterranean countries and especially widespread in Finland, where its age-standardized frequency is 1.53/100 000. 7 However, cases have been reported in other populations globally, albeit at lower frequencies. The exact global prevalence remains uncertain owing to its rarity. 8
The pathophysiology of ULD is not completely understood; however, recent research has shed light on the key mechanisms involved. Loss-of-function mutations in the EPM1 gene result in the unstable expansion of a dodecamer minisatellite repeat unit in the promoter region, which encodes CSTB, an intracellular cysteine protease inhibitor. CSTB deficiency leads to dysregulation of protease activity, subsequent cellular dysfunction, apoptosis, and oxidative stress. Loss of CSTB function is also associated with alterations in GABAergic signaling, which results in impaired inhibitory neurotransmission. This disruption in the balance between excitatory and inhibitory neurotransmission contributes to the development of myoclonus and seizures in ULD. 8
The cardinal clinical features of ULD include myoclonic jerks, tonic seizures, and progressive neurological impairment. Myoclonic jerks are frequently triggered by external stimuli such as light, noise, stress, or physical exertion. The severity of myoclonus can vary from mild, intermittent jerks to tonic seizure attacks, leading to unconsciousness and debilitating movements that significantly impact daily activities and independence. However, antiseizure medications can stabilize or even improve the myoclonus over time. Initially, neurological findings in ULD patients appear normal but may progress to include ataxia, intentional tremors, incoordination, and dysarthria. Individuals with ULD exhibit normal mental alertness with an average intelligence level within the normal range. However, a gradual decline in cognitive function is often observed over time, manifesting as memory and attention difficulties. Additional clinical features associated with ULD include sleep disturbances, such as insomnia and abnormal sleep patterns, as well as psychiatric symptoms, including anxiety and depression. These symptoms can significantly impact the quality of sleep, emotional well-being, and overall quality of life for individuals with ULD.6,9
Diagnosis of ULD involves a combination of molecular genetic testing and clinical suspicion. Neurophysiological abnormalities, such as preserved background activity on electroencephalogram (EEG) spontaneous generalized spike-wave discharges, photosensitivity, and giant somatosensory-evoked potentials, along with characteristic symptoms like involuntary myoclonic jerks and generalized tonic seizures, raise clinical suspicion. 10 Confirmation of the diagnosis usually requires identifying mutations in the CSTB gene, particularly biallelic aberrant dodecamer repeat expansions or compound heterozygosity for such expansions and sequence variants.10,11 A comprehensive clinical evaluation is also essential for an accurate diagnosis.10,11
Given the wide-ranging differential diagnosis of ULD, a thorough evaluation is crucial. During the onset phase, it is important to differentiate ULD from conditions like juvenile myoclonic epilepsy, which is characterized by partial myoclonus and diverse EEG abnormalities. 11 As the disease progresses, using key features to distinguish ULD from disorders such as juvenile ceroid-lipofuscinosis and Lafora disease becomes increasingly important, such as the absence of focal seizures, normal background EEG activity, and cognitive stability can aid in this differentiation.10,11 In the late phase, consideration of all PME etiologies, with a focus on cognitive integrity, is necessary. Additionally, essential myoclonus and familial adult-onset myoclonic epilepsy syndrome should be carefully considered, with emphasis on EEG patterns and the severity of myoclonic syndrome. 11
The cornerstone of ULD care involves symptomatic pharmaceutical and rehabilitative therapies, as well as psychosocial support. Valproic acid, a common antiepileptic medication, is effective as a first-line therapy in reducing myoclonus and the frequency of generalized seizures.11,12 Additional treatments such as piracetam and clonazepam can also be beneficial in managing symptoms. Emerging research suggests that newer antiepileptic drugs such as levetiracetam, topiramate, and zonisamide may have potential benefits. 11 However, managing ULD goes beyond symptom treatment and requires addressing the unique needs and psychosocial impairments of affected individuals. Regular physiotherapy is crucial for reducing motor deficits and improving mobility.11,12 Furthermore, customized psychosocial support, including social work and psychiatric counseling, is essential for overall well-being. The prognosis of ULD varies greatly among patients, depending on the severity of symptoms and the rate of disease progression. Optimal long-term outcomes and improved quality of life necessitate comprehensive, individualized, and multidisciplinary care. 11
Complications in ULD primarily result from the progressive nature of the disease and its impact on brain function. Myoclonus and uncontrollable seizures significantly impair functioning and lower the quality of life. 3 Close monitoring of treatment is necessary to manage potential side effects from antiepileptic drugs, which can exacerbate preexisting issues. However, with advances in comprehensive care plans and treatment techniques, the life expectancy of individuals with ULD has improved. 12
Our case reports a rare occurrence of the disease in infancy and emphasizes the necessity for the utilization of an early diagnostic technique for identification. The limitation arises from a lack of data on the specific challenges and considerations associated with the early onset of ULD. The focus on individualized treatment plans and multidisciplinary treatment emphasizes the integrated approach required for successfully controlling the illness and enhancing patients’ quality of life.
Conclusion
In conclusion, the presence of early-onset tonic seizures, along with pronounced developmental delay and cognitive impairment in the following years, especially when there is a significant family history, should raise concerns for progressive myoclonus syndromes. Genetic investigations can confirm the diagnosis, serving as a crucial step in the diagnostic process. The cornerstone of care for these syndromes involves a comprehensive approach encompassing symptomatic pharmaceutical treatments, rehabilitative therapies, and the provision of psychosocial support.
Footnotes
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
Written informed consent was obtained from the patient’s legally authorized representative(s) for participation in this study, including the use of anonymized clinical data for research and publication purposes.
Consent for Publication
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.