
Abstract
VEXAS syndrome, a myeloid-driven autoinflammatory disorder associated with somatic mutations in the UBA1 gene, was first described in 2020 and presents significant diagnostic challenges due to its complex clinical features, including hematological abnormalities and autoimmune manifestations. We describe a case involving a 64-year-old male presenting with persistent anemia, weight loss, fatigue, fever, and recurrent inflammatory symptoms. Diagnostic workup, including imaging, serology, and bone marrow biopsy, revealed characteristic findings, including myeloid hyperplasia, and vacuolization in precursor cells. Genetic testing identified a UBA1 gene mutation, solidifying the diagnosis of VEXAS syndrome. The patient responded to immunosuppressive treatment with prednisone and ruxolitinib, with significant improvement in symptoms. This case tells us the importance of considering VEXAS syndrome in patients with refractory systemic inflammation and hematological abnormalities, particularly in older males. Early recognition and genetic testing are crucial for guiding treatment decisions, as the condition is progressive, often relapsing, and requires multidisciplinary management.
Introduction
Understanding hematological malignancies poses a continual challenge for scientists. By delving into both clinical aspects and molecular pathways, tailored targeted therapies can aim for prolonged progression-free survival or even complete remission. A recent breakthrough in the field of myelodysplastic syndromes (MDSs) has shown that a single somatic mutation can be linked to both MDS and autoimmunity. 1
The VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic), a myeloid-driven autoinflammatory syndrome associated with hematological neoplasms, was first described in 2020 by Beck et al. 1 It arises from somatic mutations in the UBA1 gene located on the X-chromosome, leading to a wide array of hematological and rheumatic manifestations. This includes systemic inflammation, skin lesions, arthritis, macrocytic anemia, and thrombotic events. Managing and treating this condition require a multidisciplinary approach due to its varied and complex clinical manifestations. 2
We report a case of a male patient presenting with anemia, unexplained weight loss, and a recent history of cryptococcal pneumonia, culminating in the diagnosis of VEXAS syndrome.
Case Presentation
Our patient is a 64-year-old man who presented to the hematology clinic for evaluation of persistent anemia, weight loss, and fatigue. The patient reported fever, cough, and a significant weight loss of up to 40 kg over 10 months. He was also diagnosed with episcleritis of the left eye and chondritis of the left ear. He reported erythematous circumferential papules on his arms and legs that waxed and waned along with recurrent pain and edema in his right face. He was treated with a course of antibiotic for possible streptococcus infection with no significant improvement of his symptoms.
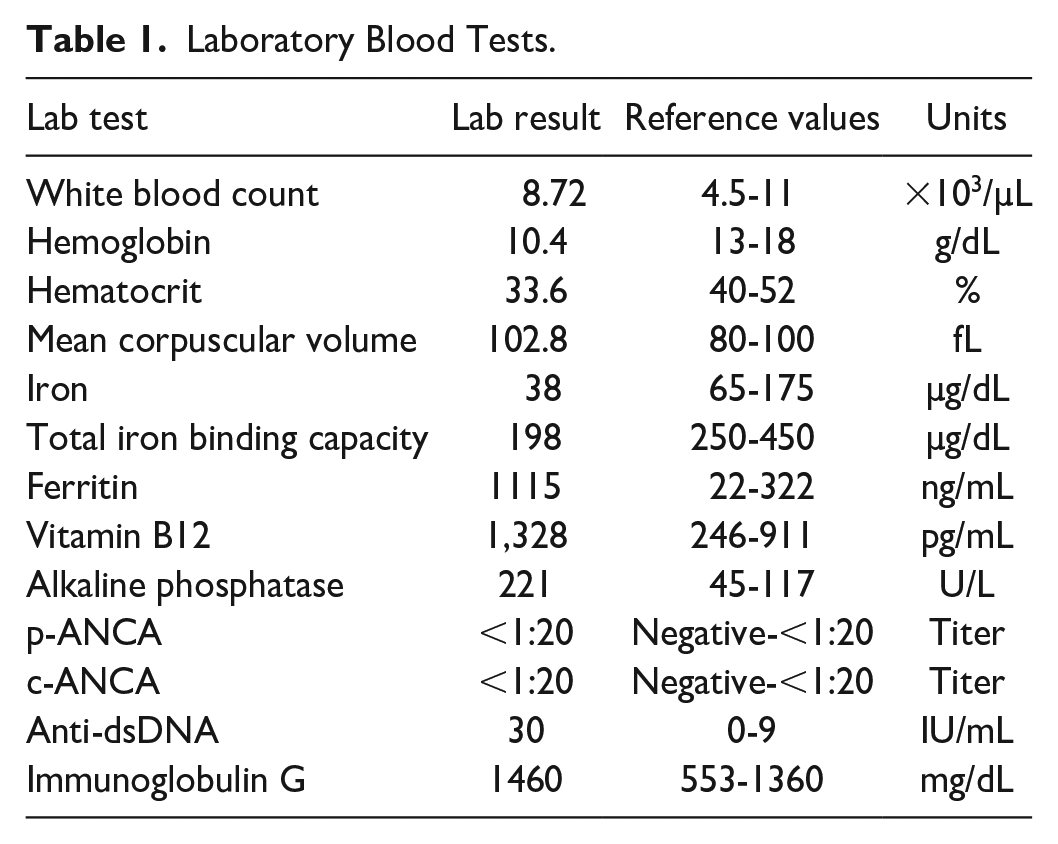
Initial blood work was significant for macrocytic anemia, with elevated alkaline phosphate (Table 1). Owing to ongoing low-grade fevers, weight loss, myalgias, and cough, the patient underwent QuantiFERON gold testing. Results were indeterminant, so the patient was referred for computed tomography (CT) scans of the chest, which revealed numerous cavitary nodules. The CT-guided biopsy of pulmonary nodules demonstrated caseating granulomas with cryptococcal organisms. Acid-fast bacillus cultures were negative; patient cryptococcal antigen levels were elevated of 1:2560. The patient was treated with a 12-month course of voriconazole without significant improvement in his symptoms. Detailed serologic evaluation for systemic autoimmune diseases was unremarkable besides a positive anti-double-stranded DNA antibody (anti-dsDNA) and positive rheumatoid factor (Table 1).
Laboratory Blood Tests.
Further workup was warranted, whole body CTs demonstrated no signs of solid tumors, and the hemoglobin electrophoresis results were normal. A bone marrow biopsy revealed hypercellular marrow with significant myeloid hyperplasia and well-demarcated small vacuoles in the cytoplasm of a few myeloid and erythroid precursors (Figure 1). Increased stainable storage iron with rare ring sideroblasts was also noted. Concurrent flow cytometry showed no immunophenotypic evidence of a lymphoproliferative disorder or an abnormal blast population. Owing to a declining hemoglobin level down to 8.7 g/dL, the patient was referred for an endoscopic ultrasound and CT enterography, both of which were unremarkable.

(A) Microscopic examination of the bone marrow core biopsy (H&E stain, 20× original magnification) reveals a hypercellular marrow for the patient’s age, with notable myeloid hyperplasia. (B) Bone marrow aspirate smears (Wright stain, 100× original magnification) demonstrate a predominance of myeloid cells at various stages of maturation. (C) High magnification of the aspirate smear (Wright stain, 1000× original magnification) highlights small, distinctive cytoplasmic vacuoles within the myeloid precursors (black arrow).
The patient presented again to the clinic for a raised, erythematous, non-blanching rash which relapses and remits every 2 weeks. A physical exam showed a rash on his bilateral upper and lower extremities, with occasional upper extremity vesicles that revealed no specific pathology when biopsied. One month later, the patient was admitted due to persistent failure to thrive despite decreasing cryptococcus titers (ratio of 1:10), increased caloric intake, and iron supplementation for anemia. Laboratory evaluation via enzyme-linked immunosorbent assay (ELISA) revealed positive double-stranded DNA antibodies (dsDNA) and positive antinuclear (ANA) antibodies, and rheumatology was consulted. Subsequent testing with gold standard immunofluorescence was negative for dsDNA and ANA antibodies. At this time, the patient was taken to Cleveland Clinic for immunology evaluation.
The patient was diagnosed with VEXAS syndrome based on a mutation of ubiquitin-like modifier activating enzyme 1 p.Met41Val (UBA1 p.M41V) detected by Sanger sequencing. He was prescribed prednisone 40 mg daily with a tapering protocol as well as 1 Bactrim 800 to 160 mg tablet 3 days a week.
Rapid improvement of his myalgias, chondritis, and rash was seen. The patient experienced profound hyperglycemia secondary to his steroids and was started on insulin. Currently, the patient is taking prednisone daily and ruxolitinib 10 mg daily.
Discussion
In this report, we outline a case of VEXAS syndrome diagnosed under our care and provide a review of the literature on this condition.
In the beginning, he presented with systemic symptoms and elevated serum levels of ferritin and alkaline phosphatase. He subsequently developed recurrent ear chondritis and ocular inflammation. In the early stages of his disease, hematological parameters were affected, leading to macrocytic anemia. The VEXAS syndrome was diagnosed based on strong clinical suspicion, laboratory findings, and the presence of vacuolization in erythroid and myeloid precursors of the bone marrow. Genetic mutations in UBA1 were confirmed, which is also necessary for diagnosis. 3 In addition, his recent cryptococcal pneumonia could be attributed to the immune dysregulation observed in VEXAS syndrome. Therapy was based on controlling inflammation using glucocorticoids.
This case aligns with the unique clinical picture of VEXAS syndrome. Typically, VEXAS syndrome is seen in males aged 60 and above. Its defining characteristics include a persistent inflammatory state alongside cytopenia and morphological vacuolization affecting myeloid and erythroid precursor cells. Despite male predominance, there are documented cases in elderly females with monosomy X, consistent with the gene’s location on the X-chromosome. 4 Within a VEXAS syndrome patient cohort, the primary disease-causing variant identified was the p.Met41Thr mutation of the UBA1 gene. Analysis suggests that mutant myeloid cells play a pivotal role in driving the inflammation seen in VEXAS syndrome, with studies indicating activation of inflammatory pathways consistent with myeloid-induced inflammation, including tumor necrosis factor (TNF), interleukin-6 (IL-6), and interferon-gamma (INF-gamma).1,5 This also suggests that myeloid precursors can persist despite UBA1 mutations, whereas mutant lymphocytes undergo negative selection in the bone marrow.
The UBA1 encodes an enzyme essential for ubiquitin activation, which plays a key role in cellular homeostasis, immune signaling, and protein degradation. 6 It has been linked to both neurodegenerative disorders and malignancies. 7 Because of its many functions, mutations in this gene can lead to a wide range of clinical presentations.
According to Beck et al, 8 since identifying UBA1 pathogenic variants can influence treatment, prognosis, and screening, broad UBA1 testing is recommended for all patients with features suggestive of VEXAS. Obiorah et al 9 outlined the primary hematologic features of VEXAS syndrome in a cohort of 18 patients, reporting macrocytic anemia in all cases (100%), thrombocytopenia in 50%, lymphopenia in 80%, monocytopenia in 50%, and neutropenia in a smaller proportion (13%). Zisapel et al 10 presented the first case of cerebral venous sinus thrombosis in a VEXAS patient.
Currently, the treatment options for VEXAS include immunosuppressive agents, including corticosteroids, Janus kinase (JAK) inhibitors, and targeted cytokine therapies. However, the efficacy of these agents remains unclear, and the treatment is considered challenging due to multiorgan involvement, variability in presentation, often accompanied by hematological malignancies, reliance on glucocorticoids, and resistance to traditional immunosuppressive treatments.11–13 Our patient is receiving ruxolitinib, which has shown superior efficacy over other JAK inhibitors according to a multicenter study by Heiblig et al. 14 This enhanced effectiveness may be attributed to its dual inhibition of JAK1 and JAK2.
Conclusion
In the coming years, general practitioners, hematologists, and rheumatologists may come across male patients exhibiting autoimmune symptoms that do not fit a typical diagnosis. Considering VEXAS syndrome as an early differential diagnosis could be appropriate in such patients. This case report brings into consideration whether an earlier diagnosis and the prompt initiation of targeted anti-inflammatory treatment might potentially improve prognosis and quality of life.
It has been nearly 5 years since the initial description of VEXAS. The overall prognosis of VEXAS syndrome remains unclear and is not yet fully defined. This may be due to its recent characterization. Nevertheless, it is considered a progressive condition that can manifest with frequent relapses and harm from both the disease and its respective treatments.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information to be published in this article.