
Abstract
Blastoid mantle cell lymphoma (B-MCL) is a rare and aggressive subtype of B-cell non-Hodgkin lymphoma characterized by a high proliferation index and poor prognosis. Gastrointestinal (GI) involvement, which is common in systemic mantle cell lymphoma (MCL), rarely presents as an isolated lesion. Herein, we describe a unique case of B-MCL presenting as an isolated cecal mass. The patient experienced acute-onset abdominal pain and melena and was found to have cecal wall thickening on imaging, with regional lymphadenopathy. Colonoscopy revealed a large ulcerated polypoid lesion in the cecum, and histopathology confirmed the diagnosis of B-MCL. The patient completed 4 cycles of bendamustine/rituximab regimen but was terminally extubated due to clinical deterioration. Although infrequent, MCL should be considered in the differential diagnosis of isolated GI masses. This case adds to the limited literature on B-MCL, which could potentially help with the formulation of diagnostic and treatment algorithms for this rare pathology.
Introduction
Blastoid mantle cell lymphoma (B-MCL) is a rare and aggressive variant of mantle cell lymphoma (MCL) characterized by medium- to large-sized lymphoid cells with a high mitotic index and a high Ki-67 proliferation index.1-4 This variant can arise de novo or as a transformation from classical MCL. 1 Blastoid mantle cell lymphoma often presents with advanced-stage disease, including frequent extranodal involvement. 5 In the gastrointestinal (GI) tract, B-MCL typically presents as multiple polypoid lesions or diffuse mucosal irregularity.6-8 Mantle cell lymphoma as an isolated cecal mass is exceedingly rare and a diagnostic challenge. 4 Management of B-MCL is challenging owing to its aggressive nature and poor response to conventional chemotherapy. 6 Current treatment regimens often result in short-lived responses, and more aggressive treatment approaches are frequently warranted.2,9 Herein, we describe an interesting case of B-MCL presenting as a solitary cecal mass in a patient with a history of a pancreatic head lesion and hypertension. 4 The patient completed 4 cycles of palliative chemotherapy and was transitioned to hospice care due to worsening functional status and increasing tumor burden.
Case Report
A 64-year-old man with a medical history of hypertension and a pancreatic head lesion (1.4 cm) presented to the emergency department (ED) with acute-onset diffuse abdominal pain and melena. The pain was dull in nature, constant, rated 9/10 at its peak, and intermittently radiated to the suprapubic area. He denied nausea, vomiting, acid reflux, acute dysphagia, hematochezia, diarrhea, or recent weight loss. In the ED, the patient appeared in mild distress due to pain. Initial vital signs were notable for a blood pressure of 88/65 mm Hg, which improved to 117/70 mm Hg after 3.5 L of normal saline boluses. On examination, the abdomen was soft and diffusely tender on light palpation but without peritoneal signs. Digital rectal examination revealed melenic stools without palpable masses, fissures, perianal ulcers, or abscesses. Triage blood tests were significant for leukocytosis (white blood cell count 11.8 × 103/mm3) and low hemoglobin (10.8 g/dL). The rest of the blood test results were unremarkable.
Computed tomography (CT) angiogram of the abdomen and pelvis was performed to rule out diverticular bleed and diverticulitis, and it showed cecal wall thickening, bilateral pelvic sidewall adenopathy, inguinal adenopathy, and enlarged lymph nodes in the peripancreatic/porta hepatis region. Carcinoembryonic antigen (CEA), carbohydrate antigen 19-9 (CA 19-9), and alpha-fetoprotein (AFP) were normal. Contrast-enhanced CT scan of the chest, abdomen, and pelvis revealed a large cecal mass measuring 6.9 × 6.0 cm, with regional lymphadenopathy. Given the history of a pancreatic head lesion and peripancreatic lymphadenopathy, magnetic resonance cholangiopancreatography (MRCP) was performed, which showed a cecal mass with extensive lymphadenopathy and several lesions in the liver suspected to be metastatic. The hepatic lesions were deemed too small for image-guided biopsy.
Esophagogastroduodenoscopy (EGD) with endoscopic ultrasound (EUS) showed diffuse pancreatic lobularity and a normal main pancreatic duct. Gastric biopsies showed focal atypical lymphoid aggregates, consistent with minimal involvement of MCL and moderate chronic inactive gastritis. The biopsies were negative for Helicobacter pylori, intestinal metaplasia, or dysplasia. Colonoscopy revealed nodular mucosa in the ascending colon and the cecum. An ulcerated and partially obstructing large mass was found in the cecum (Figure 1). Histopathology revealed MCL with a blastoid morphology and a high proliferation index (Figure 2). The atypical lymphoid cells were diffusely positive for CD20, CD5, cyclin D1, Bcl-2, and CD43 (Figure 3). CD3 staining revealed scattered, small reactive T lymphocytes. The B cells were negative for CD10, BCL6, MUM1, and TdT. Staining with c-Myc showed <30% c-Myc-positive cells. Immunostaining results for p53 were negative. Fluorescence in situ hybridization (FISH) analysis was positive for CCND1/IGH [translocation t(11;14)] gene rearrangement, supporting the diagnosis of mantle cell lymphoma.

Endoscopic images showing an ulcerated partially obstructing large mass in the cecum (panel C & D; big yellow arrows) and nodular mucosa in the ascending colon (panel A & B; small arrows).
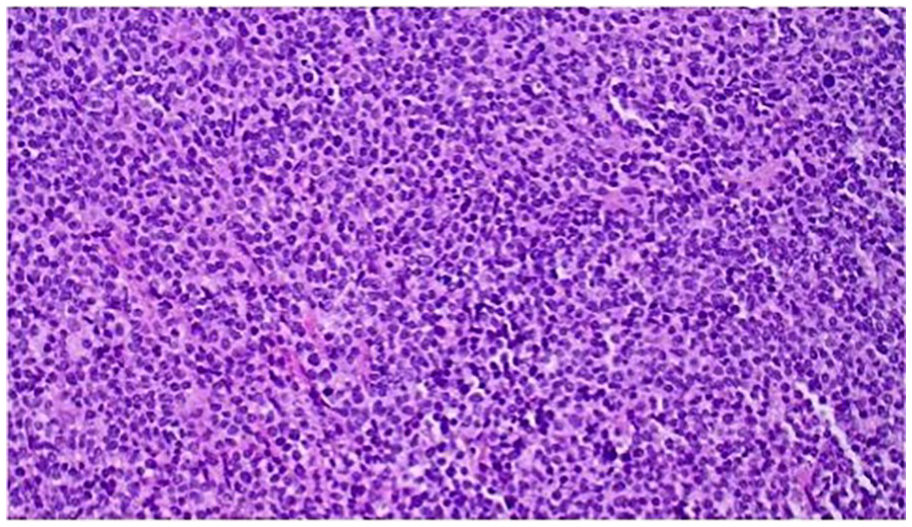
A histology slide of the cecum specimen showing diffuse monotonous atypical lymphoid infiltrates. The lymphoma cells in this specimen are mostly medium- to large-sized cells with slightly irregular nuclear contour, dispersed chromatin, and scanty to moderate amount of cytoplasm.
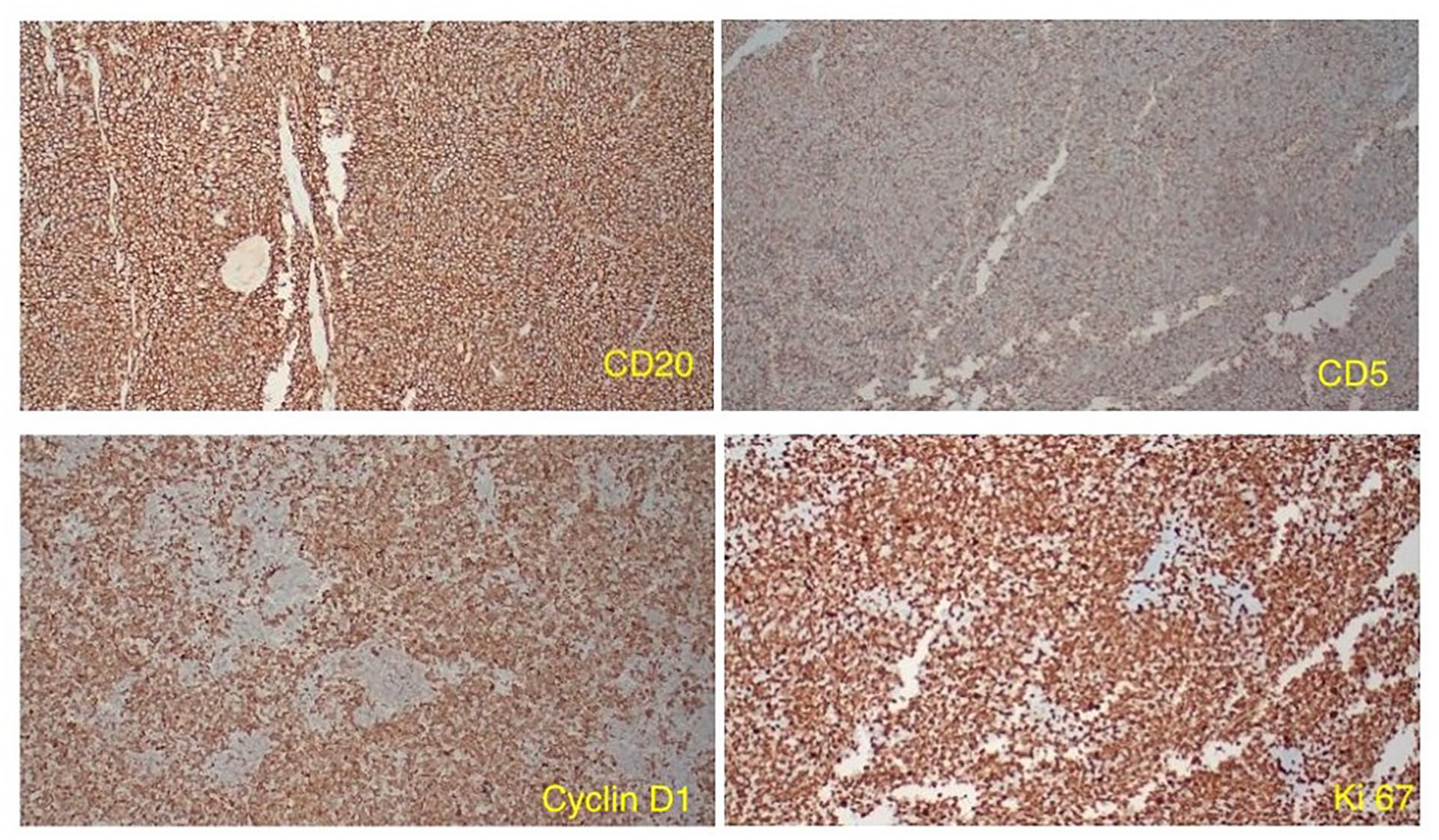
Immunostaining slide showing atypical lymphoid cells diffusely positive for CD20, CD5, Cyclin D1, and Ki-67.
The patient’s hospital course was complicated by septic shock due to Clostridioides difficile infection (CDI) and was successfully treated with oral vancomycin. After being transferred to the regular medical unit, the patient had an acute abdomen, raising concern for toxic megacolon versus ileus. A repeat CT of the abdomen and pelvis without contrast showed a large mass adjacent to the cecum, causing an obstruction near the ileocecal valve. A multidisciplinary team, including surgeons, oncologists, and the patient’s family opted for chemotherapy over surgical resection, resulting in the resolution of the obstruction. After 4 cycles of palliative chemotherapy consisting of bendamustine and rituximab (BR) as outpatient, the patient was readmitted to the hospital for septic encephalopathy, shock, and pancytopenia. Five days passed without clinical improvement, and the patient’s family opted for terminal extubation.
Discussion
In this case report, we share our experience diagnosing and managing a rare case of B-MCL presenting as an isolated cecal mass. The patient presented to the ED with acute-onset abdominal pain and melena and was found to have a cecal mass and regional lymphadenopathy on diagnostic imaging. A colonoscopy with biopsy confirmed a diagnosis of B-MCL. Mantle cell lymphoma in the GI tract usually manifests as multiple lymphomatous polyposis, making this case a rare event. 4 The patient’s hospital course was complicated by septic shock due to CDI and acute abdomen, prompting the initiation of a BR regimen for decompression. After 4 cycles of outpatient palliative chemotherapy, the patient was transitioned to comfort care due to worsening clinical status.
Blastoid mantle cell lymphoma is a rare and aggressive subtype of MCL characterized by a high proliferation rate and poor prognosis.1,10 It accounts for approximately 10% to 20% of all MCL cases. 10 The median age at diagnosis is 60 to 63 years, with a male predominance.1,6,8 Gastrointestinal involvement is typically seen at advanced stages of the disease, and the stomach is the most affected site. 5 Mantle cell lymphoma arises from naive pre-germinal center B cells in the mantle zone of lymphoid follicles. 11 Its pathogenesis involves overexpression of protein cyclin D1 located on chromosome 11 due to t(11;14) (q13; q23) translocation and a fusion of CCND and immunoglobulin heavy chain (IGH) genes.8,12-14 Blastoid variants, such as those affecting the cecum, exhibit additional genetic alterations, including mutations in TP53, ATM, or RB1, leading to genomic instability and increased aggressiveness.1,11 Gastrointestinal involvement, present in up to 90% of cases of systemic MCL, reflects the lymphoma’s tropism for mucosal sites.6,8 The molecular pathogenesis of cecal MCL encompasses both local and systemic processes. Tumor cells infiltrate the GI tract via adhesion molecule interactions, enabling migration and survival within the intestinal microenvironment. 9 This results in disruption of mucosal architecture and immune evasion. 15 Local immune dysregulation contributes to the observed mucosal thickening, ulcerations, and polypoid lesions seen endoscopically. 16
Mantle cell lymphoma of the GI tract has a spectrum of clinical features, ranging from asymptomatic to acute abdomen. Abdominal pain is one of the most common presenting symptoms, typically localized to the right lower quadrant in cases involving the cecum.2,17 This may be accompanied by changes in bowel habits, including diarrhea, constipation, or alternating patterns.3,12 Gastrointestinal bleeding, either occult or overt, is another frequent manifestation, presenting as melena, hematochezia, fatigue, or pallor.11,17 B-symptoms such as fever, night sweats, and weight loss may be observed in advanced disease stages, reflecting the lymphoma’s systemic involvement.6,12 Patients may also report nausea, vomiting, or anorexia, particularly if obstruction or severe mucosal involvement occurs. In some cases, GI MCL can cause bowel obstruction or perforation, prompting urgent intervention. 5
The diagnosis of MCL involves a comprehensive evaluation with laboratory tests, imaging studies, genetic and molecular markers, and tissue biopsies. A complete blood cell count (CBC) may reveal anemia, thrombocytopenia, or neutropenia. Elevated levels of lactate dehydrogenase (LDH) and uric acid are commonly associated with increased tumor burden and a poorer prognosis. 11 Bone marrow involvement is common in MCL cases; therefore, bone marrow biopsy is essential for diagnosis, staging, and prognostication. Bone marrow biopsies typically show extensive interstitial to diffuse infiltration. In approximately half of the cases, the blastoid variant has higher mitotic rates and a more significant proliferative index. 9 Imaging studies play a critical role in the assessment of MCL, including staging. The European Society of Medical Oncology (ESMO) guidelines recommend bone marrow biopsy and CT scans of the neck, chest, abdomen, and pelvis, as part of the initial workup for MCL. A PET-CT scan is highly recommended for the rare early stages I/II before starting localized radiotherapy. 2 Additionally, the Ki-67 index, which measures cell proliferation, is assessed to gauge the aggressiveness of the lymphoma. High Ki-67 levels are indicative of a more aggressive disease course. 14 Genetic studies are performed to identify mutations in genes such as TP53 and CCND1, which are often associated with MCL and can influence prognosis and treatment decisions.
Cerebrospinal fluid (CSF) analysis helps rule out central nervous system (CNS) involvement, which, although rare, can occur in aggressive forms of MCL. Central nervous system infiltration is diagnosed based on clinical symptoms and the identification of malignant cells in the CSF, confirming MCL. 11 Endoscopic procedures, including EGD and colonoscopy, are frequently used to detect GI involvement in patients with GI symptoms. The MCL of the GI tract can present with characteristic endoscopic features such as superficial, protruded, or ulcerative lesions in the stomach or lymphomatous polyposis from the duodenum to the rectum.7,17 Cyclin D1 is typically expressed in MCL cells and is used to confirm the diagnosis. Histopathological analysis and immunostaining are equally important for diagnosing MCL. Cyclin D1 overexpression, resulting from the t(11;14) translocation, is a hallmark of MCL and helps distinguish it from other lymphomas.2,6 The integration of these diagnostic tools provides a comprehensive approach to accurately diagnose and stage MCL, ultimately guiding effective treatment strategies.
The treatment of B-MCL is particularly challenging owing to its aggressive nature and poor prognosis. Therapeutic approaches typically involve a combination of intensive chemotherapy, targeted therapies, and stem cell transplantation. The National Comprehensive Cancer Network (NCCN) guidelines recommend stage-specific treatments, and first-line treatment often includes high-dose cytarabine regimens. 2 These regimens are preferred because of their efficacy in achieving remission in aggressive MCL variants. The rituximab, dexamethasone, cytarabine, cisplatin (R-DHA) regimen is indicated for stage 1 and 2 cancers, with R-CHOP as an alternative. For patients who are not candidates for intensive therapy or ASCT, a combination of rituximab, bendamustine, and cytarabine (R-BAC) has shown promising results.1,10,14,18 This regimen is well-tolerated and has demonstrated high overall response rates, even in older patients.
For younger, fit patients, the British Society of Hematology recommends induction therapy with rituximab and high-dose cytarabine, followed by ASCT and maintenance rituximab.2,9,11 For patients with a TP53 mutation, alternative strategies are recommended, and ibrutinib may replace ASCT during induction and maintenance if approved.11,12 For relapsed or refractory MCL cases, newer agents such as ibrutinib, lenalidomide, and venetoclax have shown significant efficacy, offering alternatives when traditional chemotherapy fails. 11 The guidelines underscore the necessity of tailoring treatment regimens to individual patient profiles, considering factors such as age, comorbidities, and response to initial therapy. 2 Surgical intervention is generally reserved for diagnostic purposes and the management of disease-related complications, such as bowel obstruction, intussusception, and perforation. 8 While not a primary treatment modality, surgery can play an essential role in the multidisciplinary management of MCL, especially in complex presentations. The median survival of MCL patients with treatment is typically 4-5 years, with worse outcomes when one or more extranodal sites are involved.6,18 The prognosis is assessed using the Mantle Cell Lymphoma International Prognostic Index (MIPI) score, which incorporates age, performance status, serum LDH level, and white blood cell count to stratify patients into different risk categories.1,10,14 This scoring system aids in predicting outcomes and guiding treatment strategies. Higher MIPI scores correlate with poorer prognosis, indicating more aggressive disease and the need for intensive treatment. 7
Conclusions
Mantle cell lymphoma of the cecum is a rare and aggressive malignancy that often presents with nonspecific GI symptoms including abdominal pain, altered bowel habits, and GI bleeding. Diagnosis relies on endoscopic evaluation, histopathological analysis, and molecular studies, with hallmark findings, such as cyclin D1 overexpression and t(11;14) translocation. Despite advancements in immunotherapy and targeted therapies, such as BTK inhibitors and CAR-T cells, the prognosis remains poor, particularly for aggressive blastoid variants. Here, we describe an interesting case of B-MCL presenting as an isolated cecal mass. After 4 cycles of palliative chemotherapy with a BR regimen, the patient was transitioned to comfort care because of worsening clinical status. This case underscores the need for early recognition, prompt intervention, and a multidisciplinary approach to improve outcomes in patients with MCL of the GI tract.
Footnotes
Author Contributions
L.B. conceptualized the idea for this case report. J.L.C., R.B., H.F., and I.D.A. assisted with the literature review, data curation, and writing of the manuscript. L.B. and Y.C. edited and proofread the manuscript. All authors have approved this manuscript for publication.
Data Availability Statement
Further inquiries can be directed to the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require IRB approval for reporting individual case reports.
Informed Consent
Verbal informed consent was obtained from the patient for his anonymized information to be published in this article.
Disclosure
This case report was presented as an abstract at the American College of Gastroenterology (ACG) Annual Scientific Meeting, 2024, in Philadelphia, Pennsylvania. Bathobakae, Lefika MD; Lopez, Jorge MD; Bashir, Rammy MD; Farhan, Heba BS; Elagami, Mohamed MD; Amer, Kamal MD; Cavanagh, Yana MD. S3114—Blastoid Mantle Cell Lymphoma Presenting as a Cecal Mass. The American Journal of Gastroenterology 119(10S):p S2112-S2113, October 2024. DOI: 10.14309/01.ajg.0001041824.81589.03.