
Abstract
Anti-glomerular basement membrane (GBM) antibody nephritis is defined by linear immunofluorescence staining of GBM by immunoglobulin G (IgG), typically associated with GBM rupture, fibrinoid necrosis, and crescent formation. Clinically, the patients present with rapidly worsening renal function, often with hematuria. Typical renal pathologic findings include necrotizing and crescentic glomerulonephritis. In contrast, thrombotic microangiopathy (TMA) is characterized by microvascular thrombosis, which can also lead to acute kidney injury. Thrombotic microangiopathy is associated with some systemic diseases and has characteristic clinical features of microangiopathic hemolytic anemia, platelet consumption, and multiple organ failure. Anti-GBM nephritis associated with TMA has rarely been reported. We describe an unusual case of atypical anti-GBM disease without crescent formation or necrosis but with light microscopic and ultrastructural features consistent with endothelial cell injury and glomerular-limited TMA.
Keywords
Introduction
Anti-glomerular basement membrane (GBM) nephritis is characterized by rapidly progressive glomerulonephritis (GN) with active crescent formation and direct immunofluorescence microscopy revealing linear staining of GBMs by immunoglobulin G (IgG). It is a rare autoimmune disease in which the anti-GBM antibodies target an antigen present in GBM, the carboxy terminus of the non-collagenous domain in the alpha-3 subunit of type IV collagen. 1 The circulating antibodies in anti-GBM nephritis are most commonly IgG. However, other immunoglobulin subtypes, such as immunoglobulin M (IgM) and immunoglobulin A (IgA), have been reported, 2 as well as rare reports of monoclonal, light chain restricted staining of various immunoglobulin subtypes.3,4 Triggering factors such as smoking, hydrocarbon exposure, and certain infections are theorized to expose specific cryptic antigens in the basement membranes of the lung and kidney, which can elicit an autoimmune response. 5 Atypical anti-GBM nephritis has been very seldom described, with linear GBM staining noted by immunofluorescence but without associated crescent formation or necrosis. 2
Thrombosis in the microvasculature and endothelial cell injury are characteristic features of thrombotic microangiopathy (TMA), which can be associated with some systemic diseases and lead to acute kidney injury. Anti-GBM nephritis associated with TMA has been rarely reported. In one reported study, the incidence of anti-GBM GN is reported as approximately 1 case per million per year, whereas the incidence of TMA is 17.5 cases per million per year. 6 We describe a rare case of atypical anti-GBM GN without crescent formation or necrosis but with associated features of a glomerular-limited TMA.
Case Report
A 74-year-old man was admitted to the hospital with chief complaints of worsening shortness of breath, abdominal and bilateral lower extremity swelling for 2 weeks, and decreased urine output. His medical and surgical history was significant for hypertension, type 2 diabetes mellitus, chronic obstructive pulmonary disease, coronary artery disease with coronary artery bypass grafting, chronic kidney disease stage 3a, secondary hyperparathyroidism, and gout. The patient was an ex-smoker, had occasional alcohol intake, and had no illicit drug use. Home medications included lisinopril 5 mg daily, furosemide 40 mg twice a day, insulin lantus and lispro, calcitriol 0.25 µg daily, febuxostat 40 mg daily, gabapentin 300 mg daily, and oxycodone-acetaminophen 10-325 mg 1 tablet every 6 hours as needed for pain. He denied any chest pain, palpitations, and dizziness. There was no history of cough, hemoptysis, or skin rash.
The patient’s vital signs on presentation were a temperature of 98.6 °F, heart rate of 72 beats per minute, respiratory rate of 19 breaths/minute, and blood pressure of 118/58 mm Hg. On physical examination, the patient was in respiratory distress, he had decreased air entry on both the lung bases and bibasal crackles were audible along with presence of mild bilateral lower extremity pitting edema. The patient’s abdomen was distended with a positive fluid thrill and shifting dullness. No skin rash was appreciated. There was no costovertebral tenderness. The rest of the physical examination was unremarkable. Laboratory data were summarized in Table 1. Other laboratory tests include urinalysis showing a specific gravity of 1.023 and pH of 5 and a urine study showing protein excretion of 500 mg/dL, glucose of 50 mg/dL, and 2+ blood. Urine microscopy revealed 50 red blood cells/high power field (HPF); the urine protein to creatinine ratio was 4.7 grams(g)/g. Liver function tests were normal. A peripheral blood smear revealed schistocytes and platelet aggregates. Other laboratory values showed elevated reticulocyte count, elevated D dimer level, normal fibrinogen level, mildly elevated prothrombin time, and mildly elevated partial thromboplastin time. Due to the renal dysfunction in the setting of hematuria and proteinuria, a renal biopsy was performed. Chest X-ray suggested evidence of pulmonary venous congestion.
Laboratory Data.

The biopsy specimen for conventional light microscopy, studied with hematoxylin and eosin, periodic acid-Schiff, periodic acid-methenamine silver, and Masson trichrome stained sections, revealed glomeruli with normal size and cellularity and mesangial expansion by matrix material, consistent with diffuse and mild nodular diabetic glomerulosclerosis (Figure 1). Superimposed on these changes were multifocal mesangiolysis and prominent glomerular capillary microaneurysm formation with associated intracapillary accumulation of leukocytes, erythrocytes, and focal fibrin. Endothelial cells were mildly reactive and swollen. Importantly, no crescents or fibrinoid necrosis was seen. Arteries displayed moderate intimal fibrosis, and arterioles showed muscular hypertrophy. No necrotizing arteritis, arteriolitis, capillary angiitis, or vascular thrombosis was present. Immunofluorescence microscopy (Figure 1) revealed strong linear staining of GBMs by IgG (3 to 4+), kappa (trace) light chain, and lambda light chain (2+). Additional direct immunofluorescence studies for IgG subclasses showed dominant staining for IgG1 (3) and IgG4 (4+), with lesser intensity staining for IgG2 (2+), a pattern and distribution similar to that noted for IgG. Staining for IgG3 was negative. Electron microscopy revealed GBMs with segmental subendothelial electron-lucent widening and basement membrane duplication with mesangial cell interposition and interposed trapping of electron-lucent flocculent material and occasional leukocytes (Figure 2). There was also prominent glomerular capillary microaneurysm formation with thinning of the basement membranes and intracapillary accumulation of leukocytes. Mesangial areas displayed segmental expansion by matrix material with segmental dissolution (mesangiolysis). Podocytes displayed extensive foot process effacement, particularly over areas of micro-aneurysmal dilatation.
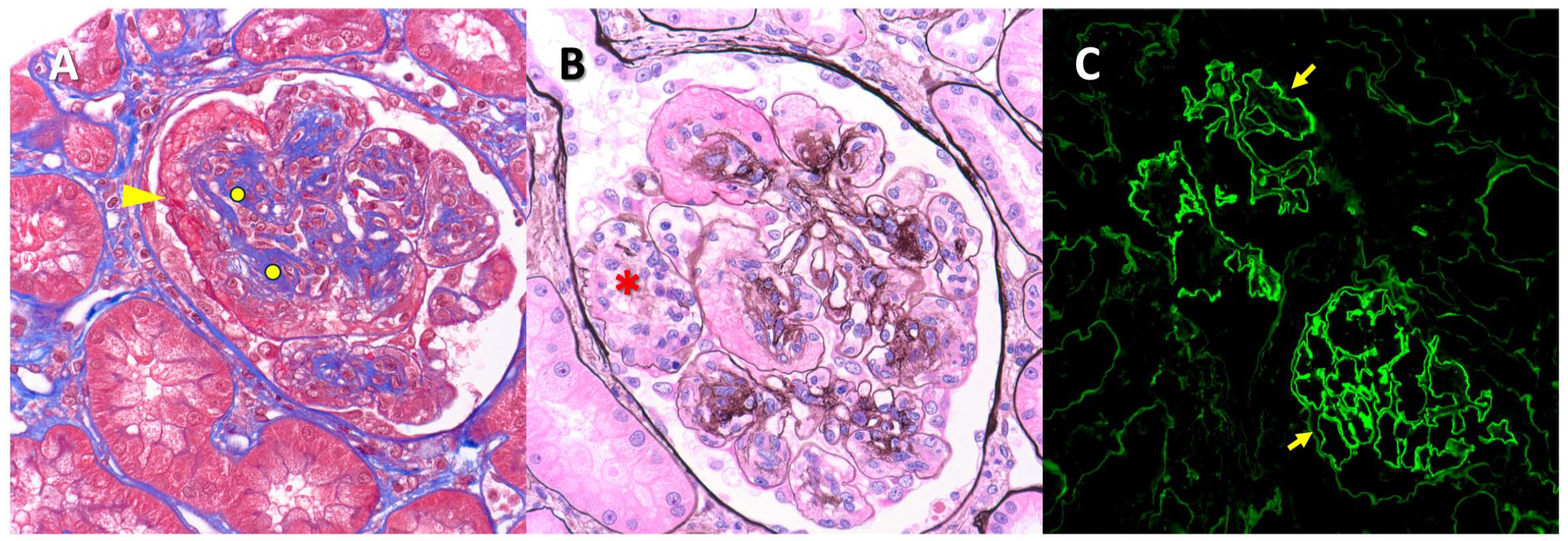
Light and immunofluorescence microscopy. (A and B) Glomeruli show diffuse and focal nodular expansion by matrix material (circle), with superimposed mesangiolysis (asterisk) and focally prominent microaneurysm formation (arrowhead) (Jones methenamine silver and trichrome stains, 400× magnification). (C) Immunofluorescence reveals strong linear staining of basement membranes (arrows), the intensity of which is considerably greater than staining of tubular basement membranes or Bowman’s capsule (200× magnification).
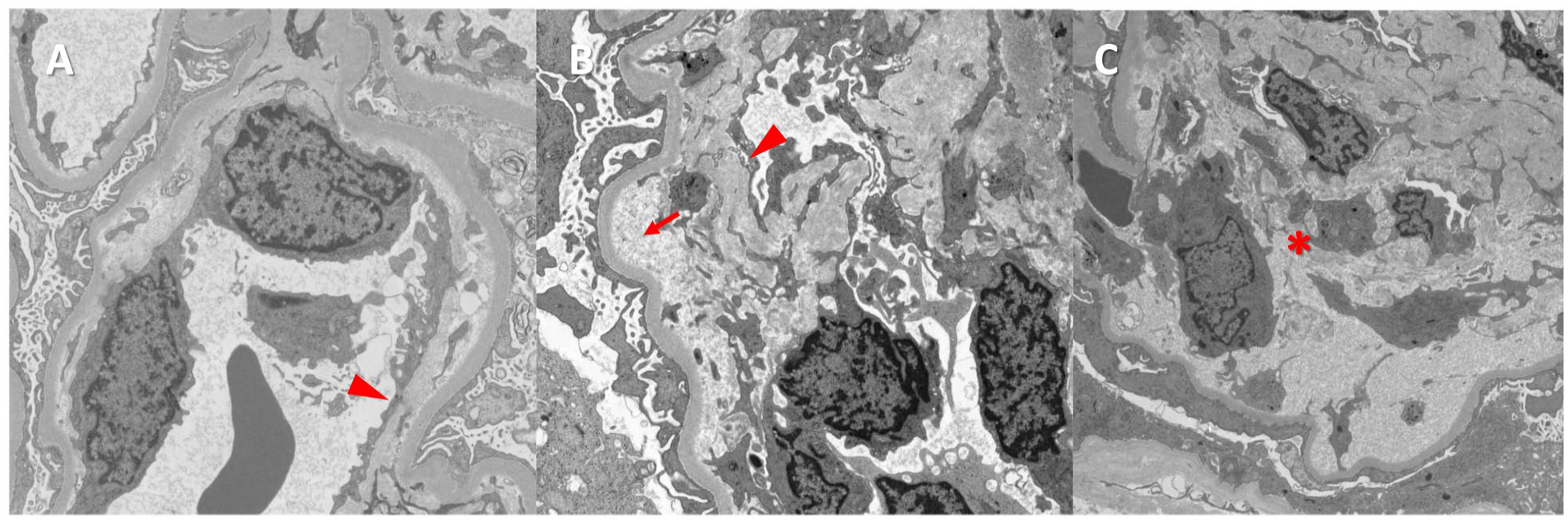
Electron microscopy and ultrastructural evaluation. (A and B) There is electron-lucent subendothelial widening with accumulation of flocculent debris (arrow) and segmental basement membrane duplication with mesangial cell interposition (arrowhead). (C) Mesangial areas show dissolution of matrix material (asterisk) with micro-aneurysmal dilation of glomerular capillary basement membranes.
Following the biopsy, additional laboratory tests were ordered. Despite the linear GBM staining by IgG noted by immunofluorescence, anti-glomerular basement antibody titers were negative. A TMA workup was also performed. ADAMTS13 activity was within normal limits, 108% (40-130), and testing for ADAMTS13 inhibitor was negative. A complete genetic panel for TMA was performed and did not identify any disease-associated mutations, including anti-complement factor H (CFH) (<1 U/mL [reference range <14 U/mL]). An autoimmune workup was also negative, including negative anti-nuclear antibody (ANA), negative lupus anticoagulant, negative anti-cardiolipin IgM (0-12), negative anti-cardiolipin IgG antibodies (0-14), IgA beta 2 glycoprotein zero U/mL (0-20), negative cardiolipin IgA levels, Immunoglobulin A phospholipid 0 unit (0-11), centromere IgG 0 AU/mL (0-40), negative double-stranded DNA, and negative scleroderma antibody IgG.
Over the course of next few days, the patient developed worsening renal function and uremic symptoms requiring initiation of hemodialysis. Although the finding of linear GBM staining by IgG on immunofluorescence was not associated with GBM rupture, necrosis, or crescent formation, the possibility that the bound IgG was causing glomerular injury manifesting as endothelial cell injury and TMA could not be excluded, and the decision was made to pursue aggressive therapy. The patient was started on a pulse dose of methylprednisolone 1 g intravenously for 3 days, followed by prednisone 60 mg daily orally and cyclophosphamide 200 mg per oral daily in the setting of atypical anti-GBM disease. The patient was also started on plasmapheresis for TMA and anti-GBM nephritis. Pneumocystis pneumonia (PCP) prophylaxis was initiated with trimethoprim-sulfamethoxazole, ceftriaxone iv was given for meningococcal prophylaxis, and the meningococcal vaccine was also administered. Because we were not noticing any substantial clinical improvement in this atypical case, the patient was also tried on iv eculizumab for 2 doses at weekly intervals. Cyclophosphamide was discontinued after 1 week because of thrombocytopenia. Unfortunately, after 3 weeks of aggressive treatment, the patient still developed multiple organ failure with renal, hepatic, and circulatory collapse and passed away after the family opted for comfort measures.
Discussion
Anti-GBM nephritis is caused by auto-antibodies directed against the non-collagenous domain of the alpha-3 subunit of type IV collagen GBMs and frequently presents clinically as rapidly progressive GN. Renal biopsy typically displays extensive glomerular crescents and necrosis associated with the characteristic linear immunofluorescence staining by IgG along the GBMs. 2 The disease is typically associated with detectable anti-GBM antibodies. However, in 2% to 3% of cases, circulating antibodies are rarely detected, and the diagnosis can only be made by renal biopsy. 7 Anti-GBM disease is a self-limiting systemic autoimmune disease that mainly affects the capillaries of the kidneys and the lungs, which share a common epitope for circulating IgG. When the disease selectively affects the kidney, it is termed renal-limited anti-GBM nephritis. The disease with concomitant renal and pulmonary manifestations, typically with hemoptysis, is termed as anti-GBM disease. Extensive crescentic glomerular involvement is common, and in active disease, the crescents tend to be in the same stage as cellular or fibrocellular crescents with or without GBM rupture and fibroid necrosis. 8 This temporal uniformity of the crescents helps distinguish it from Antineutrophilic Cytoplasmic Antibody (ANCA)-associated GN in which the crescents may display temporal heterogeneity, with cellular and fibrocellular crescents co-existing with remote, fibrous crescents, indicative of recurrent or persistent disease. 9 Any vascular inflammation in anti-GBM nephritis indicates likely an overlap with vasculitis (ANCA or non-ANCA associated).
While anti-GBM antibodies cause basement membrane injury, TMA results from endothelial cell injury, which can result in thrombosis. Compared to anti-GBM nephritis, which should not display arteriolar or arterial involvement, TMA can affect vessels of all sizes, from small to large. 10 Glomerular endothelial cell injury results in characteristic light and electron microscopic features such as endothelial cell swelling and lifting from underlying GBMs, which results in subendothelial electron-lucent widening noted by electron microscopy. Mesangiolysis or dissolution of the mesangial matrix is considered a “subacute” feature of TMA. Endothelial cell injury in arterioles and arteries can result in similar changes, such as mucoid intimal edema and fibrin thrombosis, occasionally with fragmented erythrocytes. Features of chronic TMA include GBM duplication (double contours) or concentric intimal fibrosis (“onion skinning”) in affected vessels. 11 Acute or chronic vasculopathic injury in vessels can result in downstream ischemic glomerular and tubulointerstitial injury. It should be noted that TMA should not be considered a diagnostic term but rather a pattern of injury.10,11 As such, etiology cannot be determined by pathologic features as they may appear similar or identical regardless of cause. Definitive diagnosis requires clinicopathologic correlation.
Clinically, TMA may present with thrombocytopenia, hemolytic anemia, and schistocytes in the peripheral smear; multi-system involvement can include the kidney and central nervous system. 10 TMA can be associated with systemic lupus erythematosus (SLE), antiphospholipid antibody syndrome, malignant hypertension, thrombotic thrombocytopenic purpura (TTP), typical and atypical hemolytic uremic syndrome, and pregnancy/eclampsia, among other etiologies, all of which share a common pathogenic mechanism of endothelial cell injury.10,11 Anti-GBM disease associated with TMA is sporadic and has been reported in only a few cases in the literature,1,5,6,12- 23 as in Tables 2 and 3. The exact mechanism for the overlap still needs to be elucidated.
List of Published Cases—Anti-GBM Glomerulonephritis Associated With Thrombotic Microangiopathy Renal Biopsy Findings, Treatment, Prognosis, and Mortality.

Abbreviations: TMA, thrombotic microangiopathy; RBC, red blood cell; IgG, immunoglobulin G; GBM, glomerular basement membrane; BM, basement membrane; GN, glomerulonephritis.
List of Published Cases—Anti-GBM Glomerulonephritis Associated With Thrombotic Microangiopathy Demographics and Clinical Characteristics.

Abbreviation: GBM, glomerular basement membrane; MAC, membrane attack complex.
In the patients with anti-GBM nephritis, 97% have crescents, and 85% have more than 50% glomerular involvement by crescents. 12 Not surprisingly, most of the cases reporting the coexistence of anti-GBM nephritis and TMA also had glomerular crescents. Only one case of TMA with atypical anti-GBM nephritis without crescents on renal biopsy has been reported. 12 It was postulated that occlusion of vascular lumina by microthrombi prevented the anti-GBM antibodies from reaching the GBM, thereby failing to elicit GBM injury and crescent formation. 12 Our case had a similar presentation on renal biopsy without any crescent formation. The predominant antibody involved in the pathogenesis of anti-GBM nephritis is IgG, which is detected in 95% of patients by immunoassays. 1 Positive anti-GBM titers are pivotal in confirming a diagnosis of anti-GBM nephritis. However, 2% to 3% of cases are seronegative, and in these cases, a diagnosis can only be established by renal biopsy, which can result in a significant delay in treatment. 7
In a review by Stave in 1984, the coexistence of anti-GBM nephritis and TMA was reported in 6 of 12 patients. 13 Although 2 patients did not have renal biopsy evidence of TMA, they had clinical evidence of microangiopathic hemolytic anemia. Stallworthy reported a case of anti-GBM GN with hematological evidence of TMA appearing later in the clinical course. The patient’s renal biopsy revealed active necrotizing GN with 25% crescents, but no evidence of TMA. However, the patient developed clinical features of TMA, which were initially unresponsive to plasmapheresis and had spontaneous improvement after 12 weeks. 14 In one of the reported cases, anti-GBM nephritis preceded TMA, suggesting a causal role. 12 Complement activation, von Willebrand factor levels, and genetic susceptibility may all play roles in the pathogenesis of the coexistence of anti-GBM nephritis and TMA. 15 Some reported cases had a history of smoking12,16 and hydrocarbon exposure,5,17 both of which could injure basement membranes in the lung, exposing cryptic antigens, which could then cause cross-reactive injury to GBMs.
In contrast, endothelial cell injury is the important inciting event in the pathogenesis of TMA, followed by microangiopathic hemolytic anemia, thrombocytopenia, microthrombi development, and multiple organ damage. 15 Complement activation, which is pivotal in the pathogenesis of TMA, may also play a role in anti-GBM nephritis as evidenced by the elevation of mannose-binding lectin (MBL) and complement 4a (C4a) in the serum and urine of patients with the anti-GBM nephritis. 24 Deposition of C4d is typical in anti-GBM nephritis and TMA, as demonstrated in recent studies, 24 and is caused by activation of the lectin pathway. Another possible link between anti-GBM nephritis and TMA is ADAMTS13, a metalloprotease enzyme that functions typically to cleave von Willebrand Factor (vWF). ADAMTS13 deficiency or inhibition can result in the accumulation of large von Willebrand factor multimers, resulting in abnormally sizable intravascular platelet aggregates, which can cause shear injury to endothelial cells and thrombosis. 5 There is also evidence that necrotizing and crescentic GN may also be caused by von Willebrand factor released from glomerular endothelial cells, possibly in an ADAMTS13-mediated mechanism. 15
The coexistence of anti-GBM nephritis and TMA in this patient could also be associated with genetic susceptibility, as reported by Idorn et al. 6 The case report describes pregnancy-associated TMA in the patient, but anti-GBM nephritis in the mother; the mother eventually developed end-stage renal disease (ESRD). The daughter was a heterozygous carrier of a complement I G261D mutation previously detected in patients with atypical hemolytic uremic syndrome and C3 GN. The mother, who had developed anti-GBM nephritis, did not share this mutation. However, both patients were found to have the same silent G672G polymorphism, which is strongly associated with atypical Hemolytic Uremic Syndrome (HUS) but could also be involved in developing anti-GBM nephritis. Individuals with specific genetic susceptibilities can also develop anti-GBM disease after infections, primarily mediated by the lectin pathway. 15 Vascular growth factors may also link the pathogenesis of anti-GBM nephritis and TMA. Mesnard demonstrated in an animal model with anti-GBM GN-prone mice that blocked the vascular endothelial growth factor receptor 2 (VEGFR2) signal pathway, which resulted in worsening disease severity and TMA lesions. These findings could support the common mechanistic pathway. 18
Most patients with overlap syndrome of anti-GBM nephritis and TMA were treated with immunosuppression and plasmapheresis. Immunosuppression was primarily in the form of steroids and cyclophosphamide. Vincristine was administered to one patient. 17 Our patient also received eculizumab. Almost all the patients were dialysis-dependent, and in a few cases, there was a spontaneous improvement in the hematological parameters after a few weeks. There was a poor response in most patients who had plasmapheresis. The presence of high serum creatinine, IgG1 subtypes, high anti-GBM titers, and coexistence with TMA and crescents results in End Stage Renal Disease (ESRD) and poor response to plasmapheresis.15,16
Conclusion
Anti-GBM GN is rarely associated with TMA and can result in rapidly progressive renal failure. The exact pathogenesis needs more clarification and illustration. The diagnosis is made mainly from hematological parameters, serologies, and renal biopsy. The renal prognosis is not favorable despite immunosuppression and plasmapheresis. We have described a case report of the unique overlap syndrome of atypical anti-GBM nephritis and TMA, which may help clinicians better understand and manage this unique and challenging clinical presentation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article.
Data Availability
PubMed and Google Scholar databases. The authors declare that data supporting the findings of this article are available within the article.