
Abstract
A male infant born at 34 weeks’ gestation presented with acute cardiorespiratory decompensation soon after birth followed by renal failure. Initial clinical course was complicated by ventilator requirement, bilateral pneumothoraces, and hypotension managed with multiple inotropes. Persistent renal failure with oliguria and renal ultrasound showing noncystic medical renal disease prompted further investigation. Whole-exome sequencing showed 2 pathologic mutations in the angiotensin-converting enzyme (ACE) gene, suggesting a diagnosis of renal tubular dysgenesis (RTD). Renal tubular dysgenesis is usually a fatal condition affecting the renin-angiotensin system with possible autosomal recessive inheritance. Acquired cases have been described in the setting of in utero exposure to medications such as nonsteroidal anti-inflammatory medications (NSAIDs) and ACE inhibitors. Renal tubular dysgenesis should be suspected in any neonate presenting with renal failure, refractory hypotension, ventilator requirement, hypoplastic lungs, renal ultrasound showing normal-sized echogenic noncystic kidneys with poor corticomedullary differentiation, and antenatal history significant for oligohydramnios. The overall prognosis of patients with RTD continues to improve with better ventilatory management and renal replacement therapies.
Keywords
Introduction
Renal tubular dysgenesis (RTD; OMIM: 267430) is a rare disease that starts in fetal life with defective development of the renin-angiotensin system.1-3 In the perinatal period, RTD often presents with oligohydramnios and evolves into renal insufficiency, respiratory failure, and hypotension in the neonatal period. At best, the diagnosis is made prenatally with molecular genetics. 2 More typically, the diagnosis is made postmortem by tissue pathology or genetic analysis. 4 Of the 150 cases of congenital RTD, only 13 are reported as surviving the neonatal period,5,6 highlighting poor survival.
Case Presentation
A male infant born to a 23-year-old multiparous woman via emergency cesarean section for breech presentation at 34 weeks’ gestation, weighing 2025 g (17th percentile). Rupture of membranes was approximately 65 hours prior to delivery with leakage of meconium-stained amniotic fluid. Pregnancy was complicated by limited prenatal care and a history of polysubstance abuse. Serologic testing during pregnancy was unremarkable, except for unknown gonorrhea, chlamydia, and group B streptococcus status at the time of delivery. Maternal urine toxicology was positive for tetrahydrocannabinol and methamphetamines.
In the delivery room, the infant had a weak cry and increased work of breathing that improved with continuous positive airway pressure. APGAR scores were 7 and 8 at 1 minute and 5 minutes, respectively. The infant was subsequently admitted to the neonatal intensive care unit for further management of prematurity and respiratory distress for which he was intubated and given surfactant. Chest radiographs revealed bilateral pneumothoraces that were managed with chest tube drainage (Figures 1 and 2). Sepsis evaluation was done, and he was started on empiric antibiotics. Urine toxicology screen in the baby was also positive for the same substances detected in maternal urine. His clinical condition continued to deteriorate with escalating ventilator requirements and hypotension requiring multiple inotropes. Echocardiogram and cranial ultrasound were unremarkable. He had minimal urine output and was transferred for higher level of care.

Left-sided pneumothorax on admission chest radiograph.
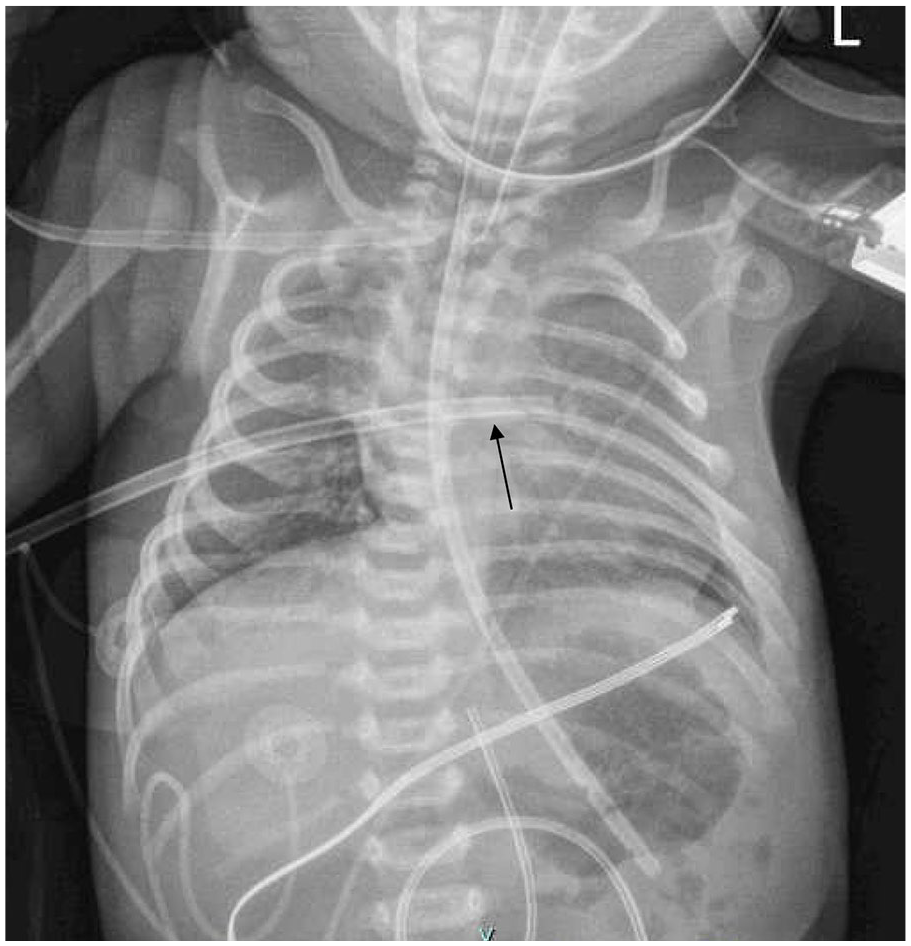
Improvement in pneumothorax after placement of chest tube (arrow).
Renal ultrasound on admission showed noncystic renal parenchymal disease with minimal urine in the bladder (Figure 3). Serum chemistry was significant for hyponatremia with a sodium of 126 mEq/L and creatinine of 3.78 mg/dL. His renal failure was managed with strict fluid balance per the nephrologist, while he remained anuric. As there was no improvement in his renal function, peritoneal dialysis was commenced on day of life 10. His cardiorespiratory status gradually improved, chest tubes were discontinued, and he was weaned off the ventilator and inotropes. Due to ongoing renal failure with persistently abnormal renal function, whole-exome sequencing was sent, which identified c. 47_70del (pLeu16_Pro23del) and c. 1522C>T (p.Arg508Ter) in the ACE gene, confirming RTD. These pathogenic mutations cause misfolding, truncation, and displacement of the ACE protein and occur with 0.0032% to 0.0096% frequency in the general population.7,8

Renal ultrasound showing noncystic parenchymal disease in structurally normal right (A) and left (B) kidneys.
Once the genetic diagnosis of renal tubular dysgenesis was confirmed, renal transplant centers were consulted. At that time, he was not deemed as a suitable candidate for renal transplant. Multidisciplinary discussions involving the infant’s family and subspecialists ensued, and a supportive care plan was formulated for him. He continued to receive peritoneal dialysis and a percutaneous gastrostomy tube (G-tube) was placed to optimize his nutrition, with the ultimate goal of better transplantation candidacy in the future. Over time, his prematurity-related issues resolved, but his renal condition showed little improvement.
At present, he is 10 months postmenstrual age (8 months corrected) and remains dialysis-dependent. He is able to feed by mouth but still requires supplementary feeds via G-tube for adequate growth and nutrition. His catch-up development is augmented with occupational and physical therapies. Although still not a transplant candidate, he receives medical care at the same quaternary children’s hospital and is being continually reassessed.
Discussion
Renal tubular dysgenesis was first described in 1983 by Allanson and has since been traditionally a histologic diagnosis, requiring renal biopsy to diagnose the specific type of dysgenesis. 9 Autosomal recessive inheritance has been suggested, with some having a family history of premature demise in the setting of oligohydramnios. Cases of acquired RTD have been described in the setting of fetal renal hypoperfusion secondary to in utero exposure to nonsteroidal anti-inflammatory medications (NSAIDs) and angiotensin-converting enzyme (ACE) inhibitors. 3 Nonsteroidal anti-inflammatory medications decrease prostaglandin concentration, leading to decreased renal vascular flow and subsequent tubular dysgenesis. 10 The ACE inhibitors can cause hypoxia, which leads to underdeveloped renal vasculature and glomerular underperfusion, causing oligohydramnios. 11 Previous study of cellular stains in tissue from patients with genetically derived RTD demonstrated the absence of proximal tubular differentiation as well as hypoplasia in the glomeruli and collecting ducts. 12 Through these mechanisms, reduced fetal glomerular filtration results in oligohydramnios. Anuria, lung hypoplasia, and refractory hypotension follow postnatally.
Recent case series compared 6 congenital RTD cases with healthy negative controls and diseased positive controls with acquired RTD from minimal change disease. 13 Immunofluorescence signals for epithelial membrane antigen (EMA) were detected in all tubular structures, but no signals were detected for CD-10 in affected tissue, specifically demonstrating the absence of proximal tubules with RTD.13,14 Patients with RTD also had increased renin expression and decreased angiotensinogen expression in hepatic tissue. 13 Although their genomic analysis detected novel mutation AGT as opposed to the common ACE mutation as in our patient, the clinical course supported what is known about fetal renal maldevelopment with RTD. All genotypic variations present similarly and require significant perinatal support with eventual need for renal replacement therapy (RRT).5,7,15,16
Similarly, refractory hypotension is universally present in RTD due to lack of a functional renin-angiotensin system. Intravenous hydrocortisone or fludrocortisone, volume replacement, and vasopressin infusion5,13 are therapies that can provide transient circulatory support. Glucocorticoids are known to stimulate transcription of angiotensinogen in the liver, and angiotensinogen II is a renal tubular growth factor. The degree of response of postnatal hypotension to steroids supports the postulation that antenatal steroids may have a role in the management of antenatally suspected RTD patients.3,13,15
In an anuric infant, antenatal oligohydramnios is an indicator of possible fetal urinary tract abnormalities. Causes of oligohydramnios can be categorized according to structure of the urinary tract. Renal agenesis/dysplasia and autosomal recessive polycystic kidney disease (ARPKD) are pathologies arising from the kidneys themselves, while obstructive uropathies typically occur within the muscular tubes of the urinary tract. Antenatal ultrasound can usually be used to differentiate these pathologies (Figure 4). Small, dysplastic, or entirely absent kidneys suggest renal agenesis, whereas large, cystic, hyperechogenic renal masses suggest ARPKD. 17 Obstruction of the urinary tract demonstrates normal/enlarged kidneys with concurrent hydroureter, hydronephrosis, and/or thickened bladder. With congenital RTD, the kidneys appear normal or slightly enlarged with echogenic noncystic parenchymal texture and have abnormal corticomedullary differentiation. As such, RTD should be suspected in any newborn presenting with profound hypotension, respiratory failure secondary to hypoplastic lungs, postnatal renal failure, noncystic, structurally normal kidneys by ultrasound, and antenatal history of oligohydramnios.

Using antenatal ultrasound to determine the location of the renal lesion.
This late preterm infant presented with acute cardiorespiratory decompensation soon after birth with bilateral pneumothoraces in the setting of premature, prolonged rupture of membranes and meconium-stained amniotic fluid. Initial differential diagnoses based on presenting symptoms included septic shock, meconium aspiration syndrome, and surfactant deficiency. Further course with renal failure, ultrasound finding of medical renal disease, and hypoplastic lungs raised the suspicion for a congenital etiology.
Renal transplantation is the definitive treatment for RTD. While the nonendocrine function of the kidney can be approximated with RRT, the endocrine functions are more difficult to manage with erythropoietin and cholecalciferol. Antihypertensive medication, electrolyte repletion, and judicious blood transfusion or iron administration often become necessary adjuncts as the disease unfolds. Options are limited in a very small infant, and peritoneal or hemodialysis can be used as a bridge to renal transplant.
Although in utero and neonatal mortality is common with RTD,1,2,10 published outcomes underestimate the survival of preterm infants with RTD13,15,18 and do not account for newer modalities of RRT that are more suitable for neonates. 5 Taken together, these palliative and curative options provide a more optimistic prognosis than expressed in some reports, 4 and this late preterm infant with postnatally diagnosed RTD is a case in point.
Footnotes
Acknowledgements
The authors acknowledge and thank the patient in this clinical report.
Authors’ Note
Some of this work was previously presented in abstract form at the Western Society for Pediatric Research Meeting, Virtual and Carmel, CA, January 22, 2022.
Author Contributions
S.G. conceptualized and designed the case report, drafted the initial manuscript, and critically reviewed and revised the manuscript for important intellectual content. P.A. conceptualized and designed the case report, and critically reviewed and revised the manuscript for important intellectual content. R.R. critically reviewed and revised the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal consent was obtained from the patient’s parents for their anonymized information for publication.