
Abstract
Nodular localized cutaneous amyloidosis is a rare form of cutaneous amyloidosis and is characterized by an extracellular deposition of insoluble amyloid fibrils which are either primarily cutaneous or a manifestation of an underlying systemic amyloidosis. Biopsy of the lesion is mandatory for the diagnosis, and histopathology shows diffuse amyloid deposits with plasmacytic infiltration. Apple-green birefringence characteristic of amyloidosis is observed when stained with Congo red and viewed under polarized light. Amyloid subtyping is done with laser microdissection followed by mass spectrometry. Majority of these lesions do not require any treatment but surgical excision, shave excision, laser therapy, and radiotherapy can be considered for symptomatic nodular localized primary cutaneous amyloidosis (NLPCA). We present a case of recurrent NLPCA in a 64-year-old woman who was treated with bortezomib and dexamethasone after failing several local therapies with excellent response.
Introduction
Nodular localized cutaneous amyloidosis is a rare form of cutaneous amyloidosis and is characterized by an extracellular deposition of insoluble amyloid fibrils which are either primarily cutaneous or a manifestation of an underlying systemic amyloidosis. Biopsy is mandatory for the diagnosis, and histopathology shows diffuse amyloid deposits with plasmacytic infiltration. Apple-green birefringence characteristic of amyloidosis is observed when stained with Congo red and viewed under polarized light. Amyloid subtyping is done with laser microdissection followed by liquid chromatography tandem mass spectrometry. Majority of these lesions do not require any treatment, but surgical excision, shave excision, laser therapy, and radiotherapy can be considered for symptomatic nodular localized primary cutaneous amyloidosis (NLPCA). We here present a case of recurrent NLPCA in a 64-year-old woman who was treated with bortezomib and dexamethasone after failing several local therapies.
Case History
A 64-year-old Caucasian woman initially presented with a skin nodule for 1 year. Her past medical history was significant for hypertension, Hashimoto’s thyroiditis, and aortic stenosis. On physical examination, a 4 × 4 cm subcutaneous skin nodule in the left shin was found. Rest of the physical examination was unremarkable and did not show any other similar nodules. Initial laboratory workup with complete blood counts (CBC) and complete metabolic profile (CMP) was normal. She underwent wide local excision and the histopathology showed abundant eosinophilic material with associated lymphoplasmacytic infiltration (Figure 1). Congo red stain also showed apple green birefringence under the polarized light. The cells were positive for amyloid P, amyloid A, but inconclusive for kappa and lambda light chains (Figure 2). Liquid chromatography tandem mass spectrometry detected the presence of AL (kappa type) amyloid deposition. These findings are consistent with amyloid tumor. Workup for plasma cell disorders was pursued with serum protein electrophoresis with immunofixation (SPEP IFE), urine protein electrophoresis with immunofixation (UPEP, IFE), serum free light chain (FLC), bone marrow biopsy and positron emission tomography (PET) scan. The SPEP, UPEP, IFE, and FLC were within normal limits. Bone marrow biopsy was also unremarkable and showed less than 5% plasma cells, and PET-CT did not show any evidence of hypermetabolic activity. Electrocardiogram (EKG) did not show any evidence of abnormal rhythm or St-T wave abnormalities. Given no evidence of systemic disease, observation was pursued. After 2 years, she noticed recurrence of the nodule in the previously excised area. Magnetic resonance imaging (MRI) confirmed the recurrence of the previous tumor (Figure 3). Re-excision was performed, and histopathology again revealed amyloid deposition. Cells were positive for amyloid P, amyloid A, but inconclusive for kappa and lambda light chains, similar to what was seen in the first lesion. Almost 1 year after her re-excision, more nodules appeared on her left lower leg. The MRI of the left leg showed at least 4 new enhancing lesions concerning for recurrence of recurrence (Figure 4). Repeat bone marrow biopsy, SPEP, UPEP, IFE, FLC, and PET-CT were done to rule out a systemic process. Bone marrow biopsy, SPEP, UPEP, IFE, and FLC were unremarkable for any evidence of plasma cell dyscrasia. The PET scan also showed a new hypermetabolic area medial to the left knee with no other hypermetabolic activity elsewhere (Figure 5). The patient did not want to pursue with further surgery and could not afford psoralen and ultraviolet A (PUVA), and therefore local radiation therapy was given to alleviate symptoms. She remained stable for a few years but started to have worsening pain and swelling in her lower extremities with new lesions on the right side. Her case was again discussed in a multidisciplinary tumor board, and it was decided to repeat the workup and start systemic treatment. Repeat bone marrow biopsy failed to show any evidence of plasma cell disorder. The PET-CT showed multiple bilateral lower extremity hypermetabolic lesions with a dominant left side lesion (Figure 6). Due to her multiple recurrences and failure to respond to local treatment, systemic treatment with bortezomib 1.3 mg/m2 on days 1, 8, and 15 in combination with dexamethasone 40 mg every week was given. Repeat PET scan after 2 cycles of bortezomib and dexamethasone showed improvement in the previous hypermetabolic lesions (Figure 7). This is a very challenging case of recurrent cutaneous amyloidosis refractory to several local treatment modalities and required systemic treatment with bortezomib and dexamethasone.

(a) Hematoxylin and eosin (H&E) stain at 4× magnification showing soft tissue mass with pale eosinophilic amorphous deposits of material with associated multinucleated giant cells (arrow). (b) H&E stain at 10× magnification showing a vessel (arrowhead) with perivascular amyloid deposition (arrow).

(a) Congo Red stain at 20× showing “apple green” birefringence characteristic of amyloid (arrow). (b) Immunohistochemistry showing staining positive for amyloid P (arrow). (c) Immunohistochemistry showing staining positive for amyloid A (arrow).

Axial view of MRI of the left lower extremity showing subcutaneous nodular lesion (red arrow) with post-contrast enhancement (blue arrow).

Axial view of MRI of the left lower extremity showing multiple subcutaneous nodular lesions (red arrow) with post-contrast enhancement (blue arrow).

(a) PET scan showing hypermetabolic uptake in bilateral lower extremities (blue arrows). (b) MIP image of PET scan identifying numerous hypermetabolic lesions in bilateral lower extremities (red arrow).

MIP image of a whole body PET scan identifying numerous hypermetabolic lesions in bilateral lower extremities, left more than right side (red arrow). No evidence of any other hypermetabolic areas was noticed.
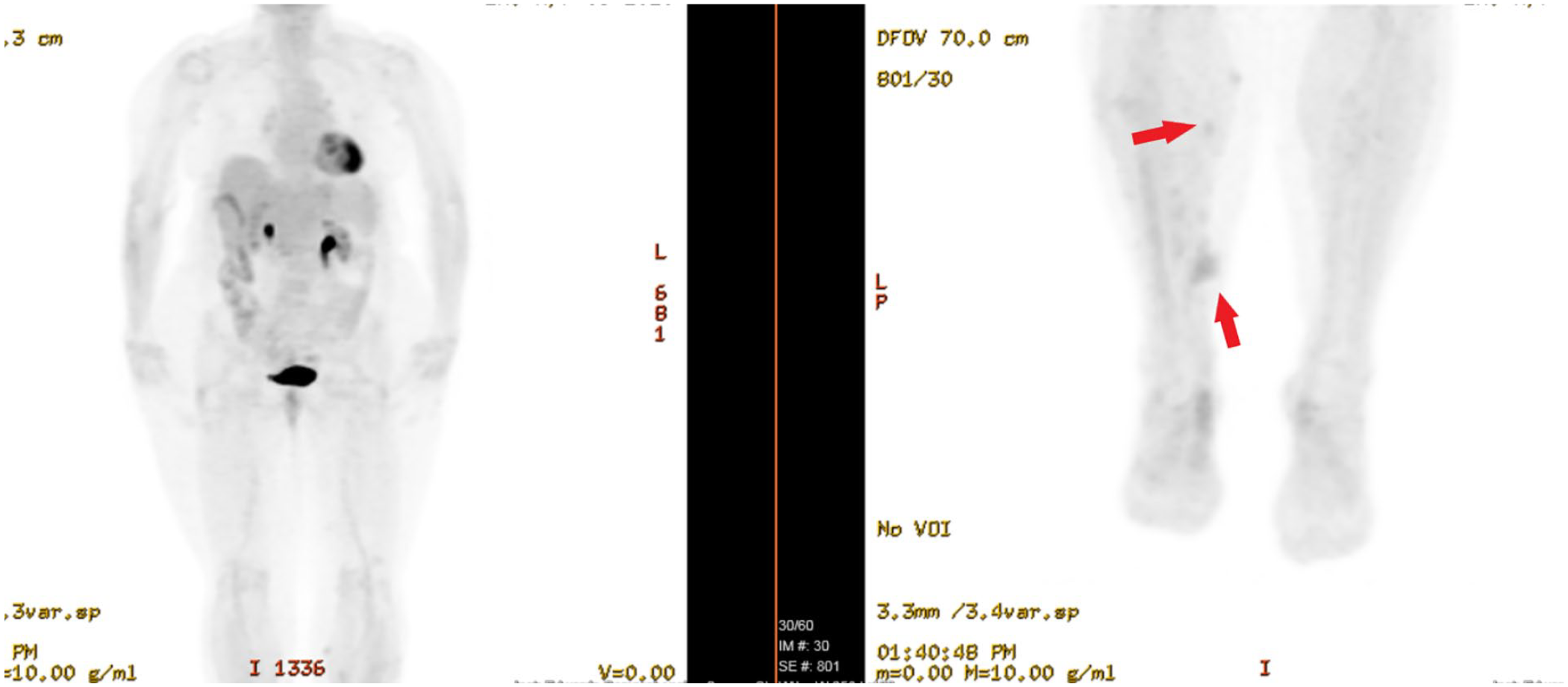
MIP image of whole body PET scan showing improvement in the previously noticed hypermetabolic lesions in bilateral lower extremities (red arrows).
Discussion
The cutaneous manifestations of amyloidosis, a spectrum of diseases characterized by the extracellular deposition of insoluble amyloid fibrils, are observed in both localized and systemic diseases. The diagnosis, and consequentially the management, of the amyloidosis subtype is grounded in hallmark clinical, histopathologic, and laboratory findings. Primary localized cutaneous amyloidosis by definition is a benign condition limited to the skin, and is subdivided into macular, lichen, and nodular amyloidosis, NLPCA. 1 The focus of this discussion, the nodular subtype, is the least common, and both its pathogenesis and presentation deviate from those described for macular and lichen amyloidosis. Behind the macular and lichen subtypes is the “keratinocyte theory,” in which degeneration of basal keratinocytes leads to the production of keratinocyte-derived amyloid and autoantibodies directed against released cytokeratin. 2 In contrast, the pathogenesis of NLPCA involves confined plasma cell dyscrasia resulting in immunoglobulin-derived amyloid, containing kappa or lambda light chains. 3
Clinical Presentation
NLPCA lesions manifest as single or multiple firm nodules, most often in the acral region, head and neck, and extremities. The plaques or waxy nodules may have overlying purpura or telangiectasia and are typically nontender. Pain, although uncommon, may result from mass effect and adjacent tissue swelling. Patients may present if the lesions are symptomatic or are of cosmetic concern and may be preceded by trauma to the affected area. 4 Concomitant autoimmune disorders are commonly associated with localized cutaneous amyloidosis. 5 Of note, up to 25% of NLPCA cases are reported with co-occurring Sjogren syndrome but the nature of this association is unclear. 6 Other autoimmune diseases, including CREST syndrome and primary biliary cirrhosis may also present alongside NLPCA. Rarely, NLPCA could be a manifestation of chronic inflammatory states, for example, long-term hemodialysis. 7
Diagnosis
The diagnostic workup of NLPCA begins with deep shave or punch skin biopsy of clinically involved sites. On histopathologic evaluation, amorphous, eosinophilic amyloid deposits can be seen extending into the dermis, subcutis, and blood vessel walls. 8 Variable levels of plasma cell infiltrate may be seen within amyloid deposits adjacent to the nearby vasculature. Apple-green birefringence characteristic of amyloidosis is observed when stained with Congo red and viewed under polarized light. The subtype of amyloid deposits must then be determined, which reveals the kappa or lambda light chain. The standard for amyloid subtyping is laser microdissection followed by liquid chromatography tandem mass spectrometry, but immunohistochemistry and immunofluorescence may be used if unavailable. 9
These histopathologic findings, however, are virtually identical to those seen in cutaneous lesions associated with systemic amyloidosis and multiple myeloma-associated amyloidosis. It is estimated that 40% of patients with systemic immunoglobulin light chain (AL) amyloidosis have cutaneous findings, which, in contrast to NLPCA, manifest in mucocutaneous regions such as the orbit and nares. 10 Diagnostic evaluation of patients with biopsy findings suggestive of cutaneous amyloidosis is therefore directed toward ruling out systemic disease. In addition to a thorough physical examination and review of systems, a CBC and comprehensive metabolic panel studies are warranted. Assessment for the presence of a monoclonal population of plasma cells outside of cutaneous lesions is achieved by serum and urine electrophoresis and immunofixation, in which the absence of monoclonal (M) protein suggests NLPCA. Serum FLC ratio analysis and bone marrow biopsy should also show the absence of monoclonal plasma cells. Urinalysis or 24-hour urine testing for proteinuria yields normal findings in NLPCA compared with systemic amyloidosis, as should imaging studies to evaluate for systemic involvement. 11 The EKG is a noninvasive and helpful tool in identifying cardiac amyloidosis and should be part of the initial workup. 12 All workup mentioned above was done in our patient and did not show any evidence of systemic disease.
Approach and Treatment Strategies
The management of NLPCA poses a unique challenge considering the relative rarity of the diagnosis and accordingly the paucity of prospective studies. An important aspect of caring for these patients involves long-standing surveillance for progression to systemic amyloidosis, of which there is an estimated 1% to 7% risk.11,13 The most commonly reported treatment for NLPCA is aimed at removal or improvement of lesion appearance. Techniques such as surgical excision, shave excision, laser therapy, radiotherapy, and cryotherapy have shown success in certain cases, but the results are inconsistent, and there is a high rate of lesion recurrence.1,2,5
In cases of treatment-resistant NLPCA, especially those compromising patients’ quality of life due to pain or disfigurement, clinicians are in uncharted territory. There have been promising applications of systemic amyloidosis-based treatment approaches in such cases 14 expanding the possibilities for case-by-case management. The limited prevalence of this condition restricts the feasibility of controlled clinical trials and the evidence-based treatment data they provide, leaving clinicians to rely on expertise and the presently available body of research. 3
While surgical and radiologic interventions are the standard for NLPCA, plasma cell-directed cytotoxic therapy is used in the management of systemic immunoglobulin light chain (AL) amyloidosis. These therapies are directed at decreasing the population of clonal plasma cells responsible for amyloid light chain production and include high-dose melphalan chemotherapy followed by autologous hematopoietic cell transplantation (HCT) or less intense outpatient chemotherapy. A regimen including bortezomib, a proteasome inhibitor, with or without corticosteroids, is a highly efficacious alternative to HCT.15,16 Options for disease that has relapsed or is refractory to initial therapy include daratumumab, an anti-CD38 monoclonal antibody, proteasome inhibitor regimens, or immunomodulatory regimens. 17 The choice of therapy is decided on according to patient-specific disease and social factors, some of which may be linked to a more favorable response to certain therapies. In particular, the immunomodulator lenalidomide with or without corticosteroids is preferred in patients initially treated with bortezomib regimens. 18
While NLPCA varies greatly in disease course when paralleled with systemic AL amyloidosis, the progress made in the management and prognosis of the systemic disease may offer an encouraging recourse for patients with refractory NLPCA.
Conclusion
The NLPCA is a rare variant of primary cutaneous amyloidosis with a small risk of progression into systemic amyloidosis. Asymptomatic patients without any evidence of systemic involvement can be clinically observed. Several local treatment modalities like surgical excision, shave excision, laser therapy, radiotherapy, and cryotherapy have shown success in cases where treatment is warranted. In cases where local modalities fail to control the disease, systemic treatment can be considered. Further studies are needed to clearly identify the role of systemic therapy in primary cutaneous amyloidosis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information including facial images to be published in this article.