
Abstract
Zirconia based ceramics are giving new hope in hard tissues replacement and implants application. Among the three forms of zirconia (ZrO2), tetragonal form (t-ZrO2) possess high mechanical stability in comparison with the other two which makes it suitable for fabricating biomedical implants with enhanced osteo activity. Here, tetragonal phase nanospheres consisting of silica stabilised zirconia (1:1) were prepared via sol gel method. The nanospheres exhibit sea urchin type morphology as observed from FESEM analysis. XRD patterns confirm the formation of t -SiO2-ZrO2 binary phase after high temperature calcination at 650°C. The immersion studies in SBF help in the formation of a layer of apatite in a gradual manner over the pallets for the period of 7, 14, 21 and 28 days which was confirmed by XRD, FTIR analysis. Moreover, t- SiO2 – ZrO2 samples were subjected to cytotoxicity tests through MTT assay on MG-63 cell lines. Antibacterial properties were investigated quantitatively using colony forming unit method against both gram positive as well as gram-negative bacteria.
Introduction
Metals and ceramics are the most widely used materials in the biomedical field. Metals like Ti, 1 Au 2 are used in bone and dental implants. The antibacterial behaviour of Au, Ag and Cu also makes these metals appropriate for dental caps. Electrodeposition is the most economical method for the production of these metallic coated dental caps.3–5 Ceramics like zirconia (ZrO2) has been extensively utilised bio inert ceramic for biomedical usage for the past four decades because of its notable features like good chemical and thermal stability, excellent osteoactivity and high hardness.6–8 However, the utilisation of ZrO2 has not been accompanied in its pure form due to the existence of three different polymorphs such as monoclinic, tetragonal and cubic at room temperature. Hence, the preparation of ZrO2 has been made by the incorporation of several metal oxides like CaO, MgO, Y2O3, Sc2O3 and CeO2 in lower concentrations in order to stabilise ZrO2 into tetragonal form (t-ZrO2) which poses high mechanical stability in comparison with other two phases.9–13
On the contrary, two major drawbacks associated with ZrO2 even after the stabilisation into tetragonal phase made its utilisation to be restricted for biomedical applications. The first drawback is the gradual transformation of t-ZrO2 to m-ZrO2 on the surface due to gradual ageing occurring over progress in time. Investigations on the ageing process of Y2O3 stabilised t-ZrO2 have been reported that diffusion of water molecules from the surrounding environment in the lattice sites of t-ZrO2 has taken place when it exposes to the body fluid at implant site followed by the volume expansion and lattice stress formation which further led to the transformation of tetragonal to monoclinic phase and formation of micro cracks at the end.9,10 A detailed case analysis presented by Scott et al. 11 shown that almost a decade after the in vivo implantation of ZrO2 there is phase transformation of t-ZrO2 to m-ZrO2 and there is a yield of around 20% wt. fraction of m-ZrO2. This phase transformation of t-ZrO2 to m-ZrO2 causes a degradation and is one of the major reason behind the weakening of the material.
Another well-known and most reported drawback of the t-ZrO2 is its poor bioactive features. Additions of bioactive ceramics such as Hydroxyapatite (HAP) and β-Tricalcium Phosphate (β -TCP) to the bio inert ZrO2 system have been attempted to prepare in the form of HAP/ZrO2 and β -TCP/ZrO2 composites for the past two decades in order to gain the dual advantages of bioactivity and mechanical stability.14–17 A large number of reports have been witnessed for the preparation of HAP/ ZrO2 and β -TCP/ZrO2 composites through either in situ synthetic approach or mechanical mixing of individually synthesised powders.18,19 The major drawbacks of those composites are the formation secondary phases such as CaZrO3, Ca3(PO4)2, Ca4(PO4)2O and CaO in addition to the presence of HAP, β –TCP, t- ZrO2 phases and phase degradation during the higher heat treatment temperatures.20,21 Due to the following mentioned circumstances, a viable substitution can be the development of completely tetragonal ZrO2 phase by incorporating an appropriate stabilising compound that has the ability to impart various required properties to the material, like good biocompatibility, no long term ageing to other phases of ZrO2 over time, and mechanical stability.
All the above-mentioned prerequisites can be achieved by the addition of silicon dioxide (SiO2) as a stabiliser. Works by Del Monte et al.22,23 has established the effect of incorporation of SiO2 for the stabilisation of tetragonal phase of Zirconia. Upon the usage of SiO2, there is an expansion in the lattice volume. This expansion in the lattice volume is due to the formation of Si-O-Zr bonds. As a result, the tetragonal phase of ZrO2 is stabilised. Numerous works in the past have shown the excellent bioactive nature of SiO2.24,25 Additionally, there are evidences that the SiO2 stabilised ZrO2 have a potential for being used for biomedical applications. Moreover, a work by Gremillard et al., have reported that by addition of small amount of SiO2 to the zirconia can avoid possible ageing of tetragonal ZrO2 and the phase transformation of the tetragonal phase to monoclinic phase. 26
By taking account on these facts, the present study aims for the development of SiO2 stabilised ZrO2 with enhanced bioactivity for biomedical applications. Various previous works have shown that the bioactive features can be introduced in a material by the presence of apatite content. In their work, Wang et al. reported that after 24 days of immersion in simulated body fluids, there was formation of an apatite layer over 12.8% calcia stabilised zirconia coatings. 27
This study aims to study the microstructural, phase stability and bioactivity behaviour of SiO2 stabilised ZrO2 after a time dependant SBF immersion. The structural characterisations of the powders are carried out using X-ray Diffraction (XRD) and Scanning Electron Microscopy (SEM) studies. FTIR, Raman spectroscopic analysis and immersion test in Simulated Body Fluid (SBF) 28 were carried out for studying the phase behaviour, vibrational modes, structural parameters, functional groups, elemental concentration, in vitro bioactivity, tetragonal to monoclinic phase transformation behaviour respectively, for the synthesised powders.
In vitro biological evaluations showed good biocompatibility of the synthesised materials with RBCs as well as Human osteosarcoma cell lines characterised by Z values, good cell viability percentage and favourable cellular morphology in the micrographs. The qualitative and quantitative antibacterial evaluation also showed slight bactericidal properties of the material enough to restrict colonisation and biofilm production upon infection.
Experimental procedure
Synthesis of SiO2 stabilised ZrO2
Sillica (SiO2) stabilised zirconia (ZrO2) is synthesised using a conventional sol-gel technique. All the chemicals used for the synthesis are procured from Sigma Aldrich (>99.99% purity). Zirconium oxychloride octahydrate [ZrOCl2·8H2O] and Tetraethyl orthosilicate [(C2H5)4OSi, TEOS] are used as a precursor for zirconia and Silica respectively. In the present study, the molar concentrations of ZrOCl2·8H2O and (C2H5)4OSi are maintained in a constant ratio of 1:1. The immersion studies are carried out in SBF solution which helps in the gradual formation of apatite layer over the pallets. The SBF was prepared in Millipore water with the recipe as described by Kokubo et al. 28 The immersion studies are carried out for the period of 7, 14, 21 and 28 days. The samples are respectively coded as SZ7, SZ14, SZ21 and SZ28, respectively and these codes are used throughout the manuscript.
The procedure for the preparation of SiO2 stabilised ZrO2 has been explained as follows. The standard solution of 1 M ZrOCl2·8H2O was prepared separately in 200 ml standard measuring flask using Millipore water. 100 ml of ethanol as a solvent was added to this mixture and this solution mixture was transferred to a 500 ml beaker and placed on a magnetic stirrer. Stirring was carried out at temperature of 80°C. An external temperature controlling probe was attached with the magnetic stirrer to maintain the temperature. The rotation speed was fixed at 350 rpm and the timer was set for 60 min of stirring. 1 M TEOS solution was then added to the solution of ZrOCl2 with continuous stirring with the same parameters as mentioned above. This was followed by the addition of 0.1 M of conc. HNO3 as a catalyst. This homogeneous solution was then slowly stirred at 150 rpm for 4 h to produce a precursor wet gel. This gel was placed in a hot air oven (First Labs) and was dried at 150°C for overnight. The dried samples were thoroughly grounded into fine powders using a planetary ball mill (tmax laboratories Ltd.) and then finally sieved by using sieve shaker (tmax laboratories Ltd.).
Structural characterisations
The phase analysis of the synthesised powders is analysed using an X-ray diffractometer (Hitachi Ltd.) with Cu Kα radiation (λ = 1.5406 nm). The radiations were produced at a voltage of 40 kV and a current of 40 mA. The samples were scanned from angles (2θ) 10° to 70° and the step size was maintained at 0.02° per second. The Raman spectroscopic analysis was employed in order to analyse the vibrational modes of ZrO2-SiO2 binary oxides powders. The powders with various days of immersion testing are analysed by XRD for studying the amount of apatite layer formed. FT-IR was carried out in transmission mode using a FT-IR spectrophotometer (Bruker Corporation). The scanning was done in the IR region (from 4000 to 400 cm−1) for functional group analysis. The powder samples were finely grounded along with a fixed amount of potassium bromide (KBr) powder, and the weight ratio was maintained at 1:100. Finally, the mixed powder was pressed into pellets. The microstructure of the powders after different days was characterised by FE-SEM combined with EDX (FEI Quanta).
Haemolysis assay
Hemocompatibility of the samples were analysed as per ASTM F 756-00 guidelines for Haemolytic assay. A test extract of the sample was prepared by suspending the sterilised samples in physiological saline for 1 h at 370°C with mild periodic agitation. After an hour the samples were allowed to rest at the bottom of container and the fluid was separated carefully to avoid any particulate matter in the test extracts using 11 μm pore size filter paper. Using a few drops of EDTA as an anticoagulant, human blood was procured from a healthy individual and suspended in a water bath at 370°C to avoid any environmental effect/stress on the RBCs. Using sterilised physiological saline, the collected blood was diluted in 1:5 ratio and this diluted blood was treated with prepared test extracts and incubated at 370°C for an hour. Distilled water was taken as +ve Control while Sterilized physiological saline was taken as −ve Control. Both the controls were also treated with 370°C temperature with the treated test extracts. After the incubation period is over, OD values of the samples and controls were recorded at 545 nm using UV Vis Scanning spectrophotometer (Shimadzu UV-1800) in triplicate numbers. The obtained values were used to calculate the Haemolysis ratio (Z) using following formula 29 :

MTT Assay [MG-63 Cells]
MG-63 human osteosarcoma cell lines with closest resemblance to the osteocytes were used to evaluate the possible toxic effects of the prepared composite in vitro. The cell lines were cultured in a mixture of DMEM, antibiotic and antimycotic solution with foetal bovine serum. Test extracts as prepared for haemolytic assay were used to evaluate the percent cytotoxicity at different release concentrations. The cells cultured in DMEM were gently rinsed with sterile PBS and incubated with 1 ml of the test extracts in 96 well plate with a serum free media at standard cell count of 1 × 104 cells/well for 12, 24, 36 and 48 h. Post incubation, the medium was aspirated from wells, treated with freshly prepared MTT in PBS and incubated again for 4 h followed by washing with fresh PBS solution. The resulting crystals were dissolved in DMSO and the absorbance change due to live cells with a change in colour from blue to purple was measured triplicately using a standard UV spectrophotometer (Shimadzu UV-1800) at 570 nm. 30 The obtained UV values were used to obtain % cytotoxicity using the following equation
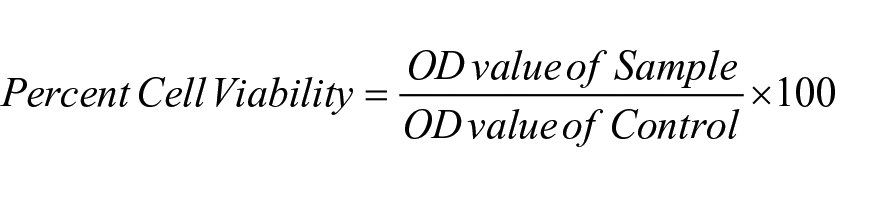
The cells were imaged using a standard optical microscope to evaluate the qualitative effects of the samples over the cell morphology with respect to the toxicity levels of the sample.
Antibacterial evaluation
Media poisoning
The test extracts as prepared for Haemolytic assay were used to prepare nutrient agar media and dried in sterilised petri plates of standard size. Standard spread plate method was employed to inoculate the serially diluted bacterial cultures over the nutrient agar medium. Carbon dioxide (CO2) atmosphere incubation of the dried petri plates was done for 24 h. The growth was imaged and analysed qualitatively to provide insights into the bactericidal properties of the material.
Colony forming units
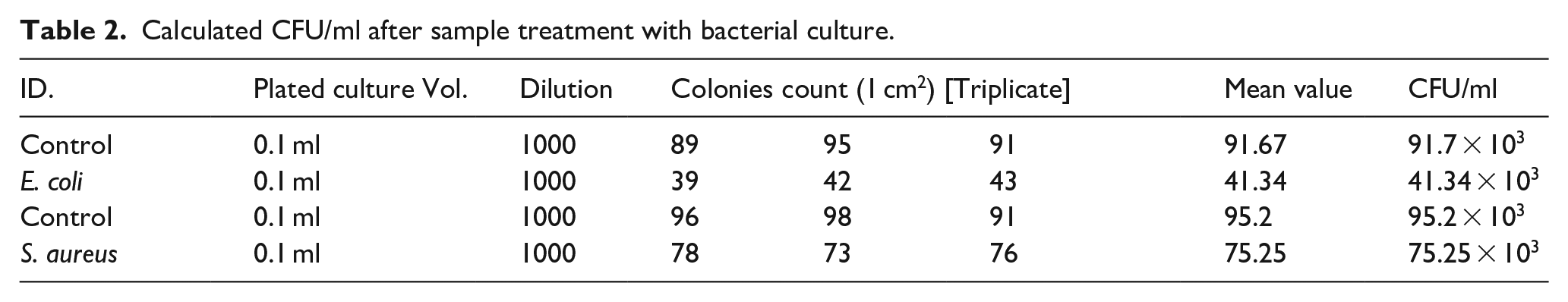
Seed culture of the respective bacteria was prepared by serially diluting the stock culture and inoculating the same in nutrient broth media followed by incubation in a CO2 incubator for 24 h. Once the seed culture was ready, it was serially diluted again to obtain the inoculation culture. 1 ml of this culture was inoculated in 5 ml of test extract and incubated at 370°C for 4 h. Unadulterated nutrient agar media was cooked, sterilised using an autoclave and solidified over standard petri plates. The test extracts inoculated with bacteria were taken and inoculated over the nutrient agar media using a standard spread plate technique. The petri plates were incubated for 24 h once inoculated. The incubation was carried out in a carbon dioxide (CO2) incubator and the colony growth was counted using a digital colony counter. Based on the triplicate counts, the CFU/ml values were obtained using the formula given below:

Results and discussion
Structural evaluation
Figure 1 shows the X ray diffractograms of the samples after SBF immersion studies after 7, 14, 21 and 28 days respectively. The indexing according to the ICDD database shows the formation of HAP after 28 days. Multiple peaks of ZrO2 and HAP are detected in the XRD spectrum. Similar results have been reported by Pessoa et al. 31 The FTIR spectrum of the samples after immersion in SBF solution is shown in Figure 2. The peaks and bands from 800 to 900 cm−1 can be attributed to silica-based groups. That intensify upon immersion in SBF due to formation of the silanol groups in vitro. The silanol groups generated courtesy of the silica-based component attracts calcium from SBF and forms silicate with the same. A further rearrangement of the compound thus generated forms amorphous calcium phosphate that eventually leads to an increase in bone mass. This unorganised bone mass eventually undergoes enzyme mediated remodelling and mineralises as new bones. 25 However, as the silanol groups are completely utilised to form HAP, the peaks decrease in intensity that confirms the active use of Silica based component of the material in formation of HAP. Also, sharp peak at around 1385 cm−1 can be attributed to phosphates which otherwise in pure phase is characteristic of Silica phase as most of the fingerprint peaks of phosphates and silica have been reported to overlap with each other. The increase in phosphate content with passage of time, and silica-based activity intensifies the peaks at ~1385 cm−1 and exploitation of silica content results in sharpening of those peaks as HAP is formed.
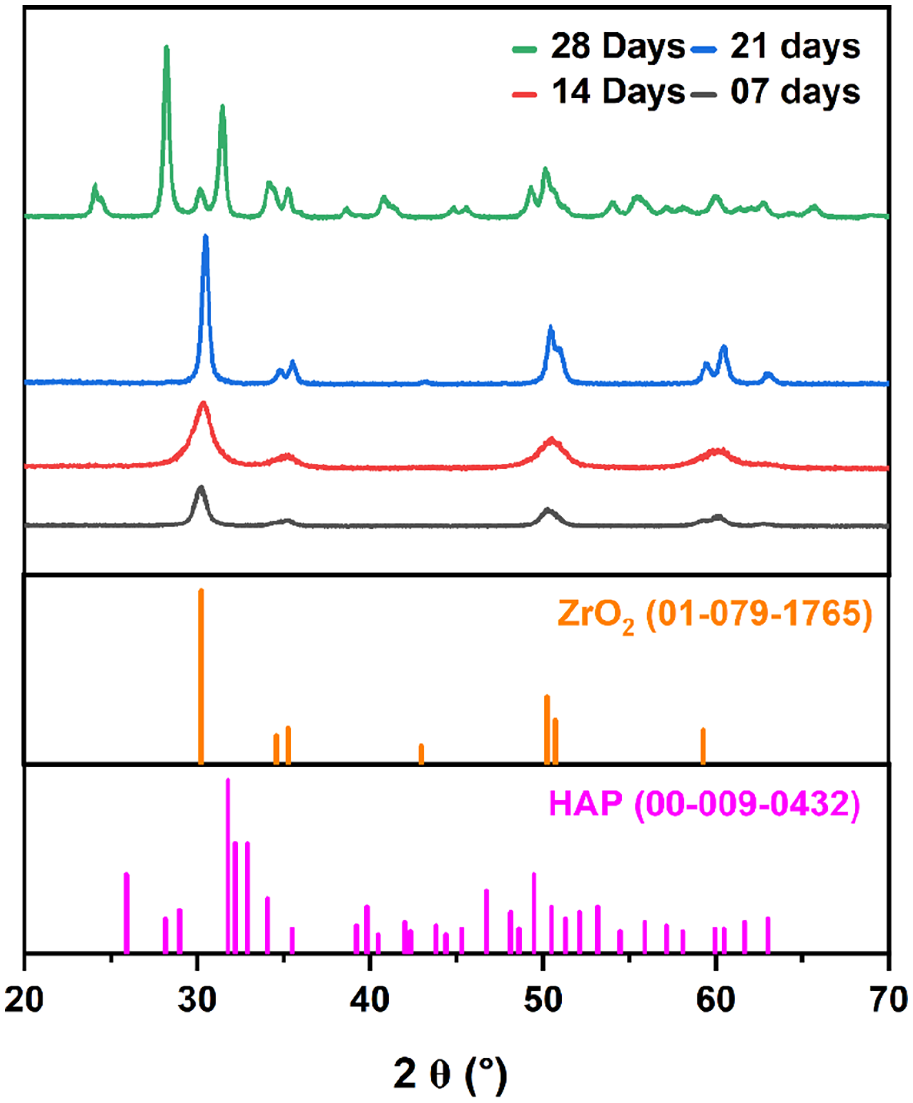
X ray diffraction patterns of the samples after immersion study in SBF solution after 7, 14, 21 and 28 days.
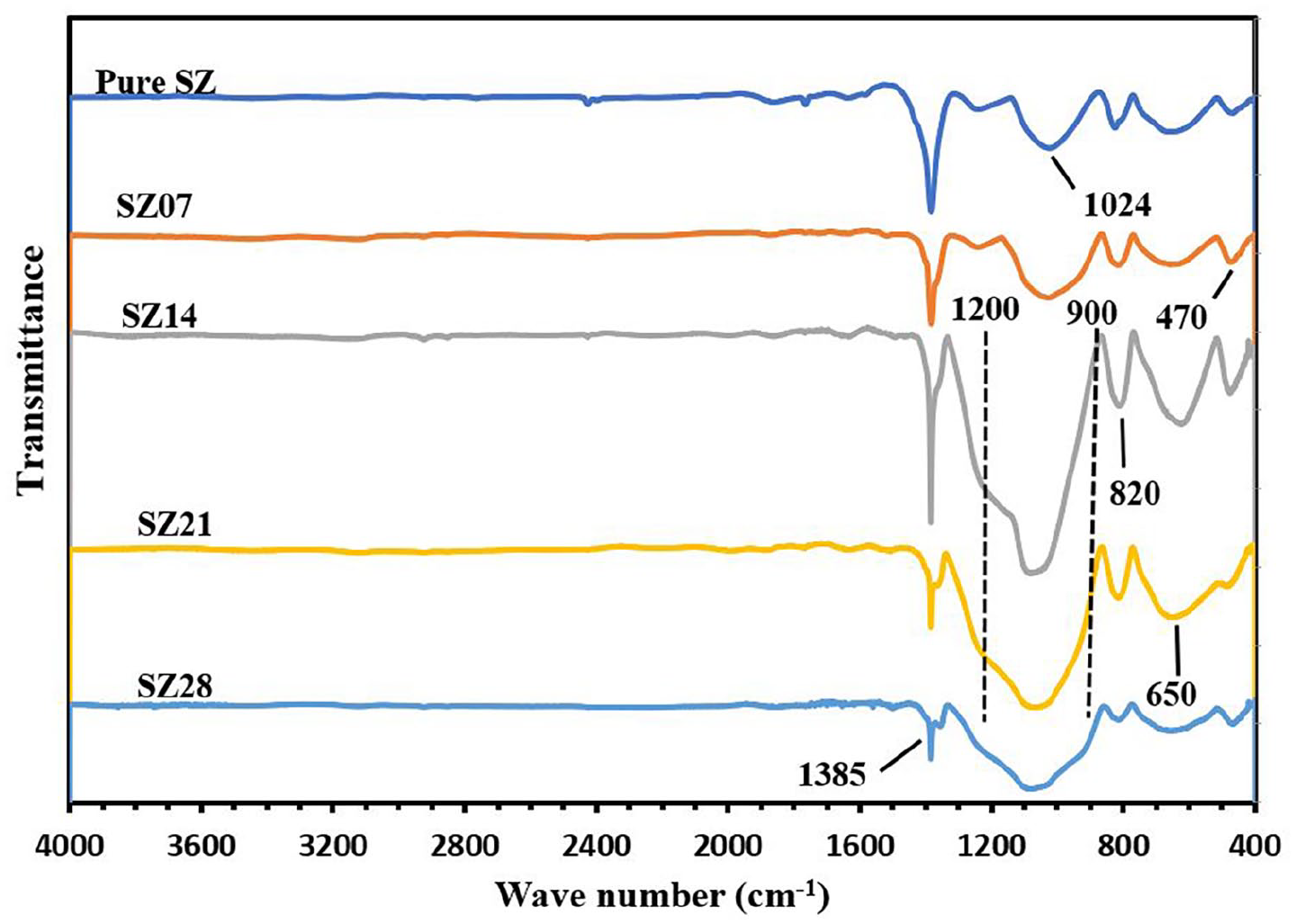
FTIR patterns of the samples after immersion study in SBF solution after 7, 14, 21 and 28 days.
The Raman spectroscopy of the samples after immersion in SBF solution is shown in Figure 3. All the peaks and bands corroborate with claims made with infra-red spectroscopy above, however, Raman spectra provides details of negligible effect over the structural integrity of the composite even after immersion in SBF after 28 days. This is a very important observation as any changes to the structural features of the composite might compromise its native features and the implantation may result in a failure.

Raman spectroscopy of the samples after immersion study in SBF solution after 7, 14, 21 and 28 days.
Hence, studies from structural characterisation clearly hints towards the involvement of the stabiliser (silica) in stimulation of bone regeneration and successful formation of HAP after 28 days of immersion in SBF in vitro. Preservation of the characteristic spectroscopic peaks and bands of zirconia shows negligible effect on the same and confirms that all the excellent and proven mechanical properties of zirconia can be exploited in long term implantation plans. Such incorporation of a bioactive phase stabilising the zirconia ceramic not only adds to its bioactivity but also preserves its native features.
The SEM micrographs of the as-prepared samples and samples after immersion testing in SBF solution for 7, 14 and 28 days are shown in Figures 4 and 5, respectively. Well distinguished crystalline morphology of zirconia crystals can be clearly observed in Figure 5(a) and (b) even after 7 days of immersion. This is a direct callback to the inert nature of zirconia that hardly allows it to interact with the SBF. Formation of amorphous calcium phosphate is clearly visible over the zirconia crystals in Figure 5(c) and (d) after respective days of immersion that eventually can grow into HAP. This deposition is caused due to activation of silica groups present within the ceramic. Silica groups acting as a stabiliser are hard to reach in such zirconia systems which is the main reason why amorphous calcium phosphate formation took almost 14 days to be actively involved in the same. The samples, after 28 days of immersion testing in SBF solution, show a typical sea urchin type morphology as shown in Figure 5(f). There is a gradual formation of apatite layer over the SiO2 stabilised ZrO2 characterised by needle shaped morphology of the newly formed HAP specific to P63/m space group of the apatite superfamily having hexagonal bipyramidal structure. This sea urchin type of morphology helps in enhancing the bio activity of the samples and hence the osteoconductivity due to an increase in surface area of interaction.

SEM micrographs of the as-prepared SiO2 stabilised ZrO2 nanospheres (a) lower and (b) higher magnifications.

SEM images showing the surface morphology of the samples after immersion test in SBF solution after 7 (a and b), 14 (c and d) and 28 (e and f) days.
Biocompatibility evaluation
The calculated values (CFU/ml) after sample treatment with bacterial culture is shown in Table 2. The haemolytic ratio calculated Table 1 as described above shows proper haemocompatibility of the samples at all of the concentration with haemolytic ratio falling way below 2 units that is the upper limit of haemocompatibility beyond which the samples turn slightly haemolytic. 29 This slight haemolytic properties if found may be acceptable in some cases but in vivo, the level of even slight haemolysis may cause issues with the implant in long term due to its adverse effects on RBC cells. 32
Calculated haemolysis ratio (Z) after sample treatment with RBCs.

Calculated CFU/ml after sample treatment with bacterial culture.
Qualitative evaluation of the RBCs (Figure 6) after treatment with sample test extracts also shows minimal effects on the structure and morphology of the cells. Figure 6 shows morphology of RBCs at (a)10 µg/ml, (b) at 25 µg/ml, (c) at 50 µg/ml and (d) at 100 µg/ml test extracts. However, due to a little stress generated by silica based component of the composite, the cells tend to agglomerate which has been reported to subside with passing time. 33 Hence, both the qualitative and quantitative evaluation of the sample’s toxicity over RBCs clears it for bone regeneration applications without any adverse effect. Cytotoxicity evaluation over MG-63 cell lines also provides similar results even after 48 h of incubation with the sample test extracts. Such promising results with high cell viability percentage (~85%) at all release concentrations shows the sample biocompatible with osteocytes that is important to promote bone regeneration (Figure 7). Qualitative assessment of the cells with micrographs also corroborates with the numerical data with promising cell viability percentage. Figure 8(a) to (d) shows morphology of cells at 10 µg/ml for 12, 24, 36 and 48 h respectively. Similarly, Figure 8(e) to (h) shows morphology of cells at 25 µg/ml release concentration. Figure 8(i) to (l) shows the morphology at 50 µg/ml and the rest at 100 µg/ml. The structural and morphological details of the samples were evaluated which shows negligible effects over the same even after 48 h at 100 µg/ml.

Optical micrographs of RBCs at (a)10 μg/ml, (b) 25 μg/ml, (c) 50 μg/ml and (d) 100 μg/ml.
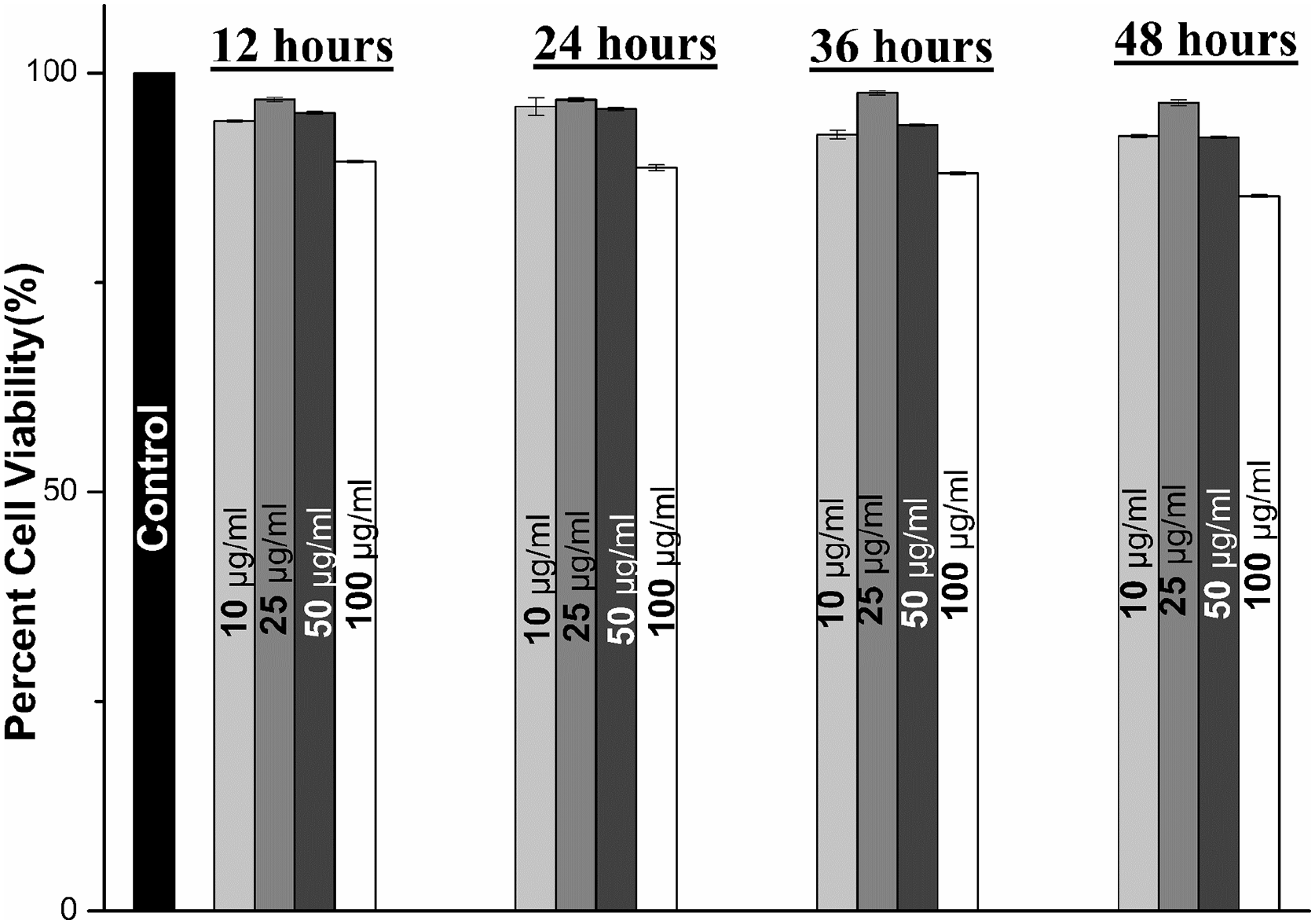
Percent toxicity of samples at different concentrations incubated for 12, 24, 36 and 48 h.

Optical micrographs of MG-63s at 10 μg/ml, 25 μg/ml, 50 μg/ml and 100 μg/ml incubated for (a-d) 12 hours, (e-h) 24 hours, (i-l) 36 hours and (m-p) 48 hours.
Antibacterial evaluation
Media poisoning method uses the antibacterial properties generated by the nutrient agar medium that is responsible for growth and metabolism of the pathogens. The parts and release products of the material restricts bacterial growth when ingested by the bacterial cell. It is clearly visible that the samples at 50 µg/ml release concentration, often considered as the most favourable for bone regeneration studies exhibits slight antibacterial properties. However small, this level of bactericidal properties is often enough to restrict bacterial colonisation and biofilm formation. It is also remarkable that antibacterial properties are higher over E. coli (Figure 9(a) and (b)) due to absence of a thicker peptidoglycan layer in cell wall that restricts the bactericidal properties of a material. 34

Optical photographs of bacterial growth over (a and b) media poisoning assay and (c and d) over CFU/ml assay.
The above trend of antibacterial properties was quantised by calculating CFU/ml values with help of calculating the grown bacterial colonies after treatment with test extracts. Similar results were obtained as compared to that with media poisoning but unadultered nutrient media allows unrestricted growth and slight aggressive colonisation is obtained (Figure 9(c) and (d)) as compared with growth of media poisoning evaluation. 35
Hence, with in vitro bioactivity evaluation of the samples, it is clear that the Zirconia-Silica composite is biocompatible, has least cytotoxic effects over native cells of humans and exhibits a respectable level of bactericidal properties that makes it a promising candidate for bone regeneration applications.
Conclusions
The structural evaluation of the proposed material shows formation of HAP with assistance from stabiliser (silica) over a prolonged period of time. Spectroscopic data also confirms negligible effect over zirconia that rules out any kind of damage to the implant in vivo. It was observed that formed hydroxyapatite over zirconia was initiated around 14 days of immersion in form of amorphous calcium phosphates. Quantitative data shows the samples are hemocompatible and biocompatible to MG-63 human osteosarcoma cell lines even at 100 ug/ml release concentration which makes them fit for in vivo implantation. Quantitative data also shows minimal effect over the integrity both RBCs as well as MG-63 cell lines. Both qualitative and quantitative antibaterial evaluation shows slight antibacterial properties of the material due to silica which is enough to prevent bacterial colonisation. Hence the proposed material is a good candidate to be deployed as an implant with great mechanical strength and induced bioactivity for prolonged periods.
Footnotes
Acknowledgements
The authors would like to thank Tongji Hospital (affiliated with Tongji University School of Medicine) and Shanghai Ninth People’s Hospital (affiliated with Shanghai Jiao Tong University School of Medicine) for providing laboratory space and research facilities.
Author contributions
JH and SL contirbuted equally to this work by carrying out the experiments and conducting the bioanalysis of the data. SA and DW derived the work concept, designed the experiments and arranged for funding to support the research work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Fundamental Research Funds for the Central Universities (grant 22120210569), the National Scientific Foundation of China (82171993), the Clinical Research Plan of SHDC (SHDC2020CR3083B), the Technology Project of Shanghai Science and Technology Commission (19441902700), the Clinical Research Program of Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYLJ202122), the Project of Biobank from Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (YBKB202116).
Guarantor
DW