
Abstract
Becker Muscular Dystrophy (BMD) is caused by in-frame mutations in the DMD gene, leading to the production of internally truncated but partially functional dystrophin. Although cardiac involvement is a major contributor to disease burden in BMD, the cellular mechanisms driving cardiomyopathy remain incompletely understood. While emerging evidence suggests that iron imbalance may contribute to oxidative stress and mitochondrial dysfunction in muscular dystrophies, its role in BMD-associated cardiomyopathy has not been defined. Building on our previous findings of dysregulated iron homeostasis in dystrophin-deficient cardiomyocytes from Duchenne muscular dystrophy (DMD), we investigated whether similar alterations are present in BMD using patient-specific and genome-corrected hiPSC-CM models. HiPSC lines derived from two BMD patients and their CRISPR/Cas9-corrected isogenic controls displayed normal karyotype, pluripotency, and efficient differentiation into cardiomyocytes (hiPSC-CMs). BMD hiPSC-CMs showed elevated ROS levels and decreased cytoplasmic and mitochondrial labile iron pools, accompanied by reduced expression of mitoNEET (CISD1), a regulator of mitochondrial iron handling. We also detected changes in the expression of genes involved in iron storage (FTH1), uptake (TFRC), and export (SLC40A1), suggesting a dysregulation of iron trafficking. Importantly, correction of DMD mutation by CRISPR/Cas9 gene editing reversed the effects observed in BMD cardiomyocytes. These results extend our previous observations in DMD to BMD cardiomyocytes and suggest that full-length dystrophin is essential for maintaining cardiac iron homeostasis.
Highlights
• Patient-specific and CRISPR/Cas9-corrected isogenic hiPSC-CMs can be used to model BMD • BMD hiPSC-CMs exhibit elevated ROS levels and reduced cytoplasmic and mitochondrial labile iron pools • Iron dysregulation is associated with reduced mitoNEET level • Restored dystrophin expression contributes to maintaining cardiac iron homeostasis
Introduction
Becker Muscular Dystrophy (BMD), which affects approximately 1 in 18,000 males, is an X-linked recessive neuromuscular disorder, typically caused by in-frame mutations in the dystrophin (DMD) gene, leading to production of a truncated, partially functional dystrophin protein. 1 By contrast, nonsense and frameshift mutations in the DMD gene cause the more severe Duchenne muscular dystrophy (DMD), characterized by nonfunctional dystrophin or, in most cases, its complete absence. 2 BMD shows a highly variable clinical presentation, from asymptomatic individuals to patients with early-onset weakness and cardiomyopathy. Many BMD patients experience significant motor impairments, approximately 60–75% develop cardiac dysfunction, with a mean age at onset of 28.7 ± 7.1 years, and up to 50% progress to overt cardiomyopathy. 1 Although severe dilated cardiomyopathy (DCM) before the age of 20 is uncommon, progressive cardiac impairment remains a major determinant of disease prognosis, often culminating in heart failure. 1
Despite the clinical relevance of cardiac complications in BMD, their molecular mechanism and potential therapeutic targets remain significantly unexplored compared to DMD. Recent studies have demonstrated that in-frame mutations, similar to those found in BMD patients, can be reproduced in animal models,3–5 enabling a more precise investigation of disease pathogenesis, including its cardiac phenotypes. Patient-specific human induced pluripotent stem cells (hiPSCs) have emerged as a powerful tool for modeling BMD, allowing the generation of cardiomyocytes (hiPSC-CMs) carrying patient-specific mutations and their functional consequences. Although no study has yet provided a comprehensive hiPSC-CMs-based model of cardiomyopathy in BMD,6,7 including ours, 8 several groups have reported the derivation and characterization of BMD patient-specific hiPSC lines.6–8 Notably, Kameda et al. 9 generated hiPSC-CMs from a female BMD carrier with a Δ45–48 DMD mutation and demonstrated that truncated dystrophin impairs contractile function in engineered cardiac tissues.
Iron is crucial for cardiomyocytes, with key roles in oxygen transport, energy production, and Fe/S cluster formation. 10 Cardiomyocyte iron homeostasis relies on coordinated activity of key proteins regulating iron import, storage, and export. 11 Iron is delivered via transferrin (TF) and imported through transferrin receptor 1 (TFRC), which is essential for cardiac function. Excess intracellular iron is sequestered by ferritin, a heteropolymeric complex composed of heavy (FTH1) and light (FTL) subunits, while iron efflux is mediated by the transmembrane exporter ferroportin (SLC40A1). 11
Recently, we demonstrated that dystrophin-deficient hiPSC-CMs exhibit significant dysregulation of iron homeostasis, including increased expression of TFRC and FTH1, and downregulation of SLC40A1. 12 Notably, we also observed a marked downregulation of mitoNEET, a key mitochondrial iron regulator encoded by CISD1, linking dystrophin deficiency to mitochondrial iron accumulation and oxidative stress. 12 However, it remains unknown whether similar disturbances occur in BMD cardiomyocytes, in which dystrophin is truncated rather than completely absent. Interestingly, among all murine tissues, CISD1 shows the highest expression in the heart. 13 C57BL/6J mice with CISD1 deletion develop heart failure and exhibit elevated levels of reactive oxygen species (ROS). 14 Moreover, the mitochondrial iron content in heart tissue is significantly higher in CISD1-deficient mice compared to controls. 14
In this study, we examined whether truncated dystrophin contributes to dysregulation of iron homeostasis in BMD by using hiPSC-CMs from two BMD patients carrying in-frame deletions (exons 45–47 and 3–9).
Materials and methods
hiPSC culture
Peripheral blood leukocytes were collected from two BMD patients treated at the Centre for Rare Diseases, Medical University of Gdańsk, following written informed consent (Approval No. NKBBN/402/2020). HiPSCs were generated from peripheral blood mononuclear cells (PBMCs) using CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific) following the manufacturer’s instructions, as previously described. 15 The first patient (BMD1) carried an in-frame deletion spanning exons 45–47 of the DMD gene, while the second patient (BMD2) harbored an in-frame deletion spanning exons 3–9. A detailed characterization of the BMD2 patient-derived hiPSC line and the corresponding CRISPR/Cas9-corrected isogenic cells has been described in our previous publication. 8 HiPSCs were cultured on GeltrexTM-coated plates (Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix, Thermo Fisher Scientific) in Essential 8 ™ (E8) medium (Thermo Fisher Scientific). After each passage (with 0.5 mM EDTA), cells were cultured in E8 medium supplemented with 10 μM Y-27632 (MCE) for 24 h. Cell were cultured at 37˚C, 5% CO2, 20% O2.
Design and construction of plasmids with sgRNAs
To facilitate correction of the DMD gene mutation, sgRNA sequences (Supplementary Table 1) were designed using the CHOPCHOP and cloned into the pSpCas9(BB)-2A-Puro vector (Addgene #62988) following the protocol of the Zhang lab at MIT. 16 The pSpCas9(BB)-2A-Puro vector was digested with BbsI (New England Biolabs, NEB) and the linearized plasmid was purified from a 1% agarose gel using the Zymoclean Gel DNA Recovery Kit (Zymo Research) according to the manufacturer’s protocol. Complementary oligonucleotides were phosphorylated and annealed in a single reaction using T4 Ligation Buffer (NEB) and T4 polynucleotide kinase (PNK, NEB) with thermocycling (37 °C for 30 min, 95 °C for 5 min, gradual cooling to 25 °C at 5 °C/min). Annealed oligos were ligated into the BbsI-digested vector using Quick Ligase (NEB) at 37 °C for 1 h. Ligation mixtures were transformed into E. coli Stbl3 cells (Thermo Fisher Scientific) via heat shock. After recovery in SOC medium, cells were plated on LB agar (BioShop) containing ampicillin (0.1 mg/ml, Sigma-Aldrich) and incubated overnight at 37 °C. Individual colonies were cultured in LB-ampicillin medium, and plasmids were isolated using ZymoPURE Miniprep (Zymo Research) and Syngen Midi kits, according to the manufacturers’ protocols. A single bacterial colony was picked and inoculated into liquid LB medium supplemented with ampicillin. The culture was incubated overnight at 37°C with shaking. To verify the presence of the plasmid containing the repair template sequence in the transformed cells, plasmids were isolated using the ZymoPURE Plasmid Miniprep Kit (Zymo Research) following the manufacturer’s protocol. Insert presence was confirmed by BbsI digestion and 1% agarose gel electrophoresis. DNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). The identity and sequence of sgRNA inserts were verified by Sanger sequencing (Genomed S.A., Warsaw, Poland) using a U6 promoter-specific primer (5′-CAGCACAAAAGGAAACTCACC–3′).
Nucleofection
HiPSCs were nucleofected with plasmids using the Human Stem Cell Nucleofector Kit 1 (Lonza) and the Nucleofector Device (program B-016). The nucleofection solution was prepared by mixing Nucleofector Solution with Supplement 1 and pre-incubating the mixture at 37 °C. In parallel, a 12-well plate was coated with Geltrex™ (Thermo Fisher Scientific). Cells were dissociated and resuspended in PBS containing 10 μM Y-27632 (PeproTech), counted, and 5 × 105 cells were pelleted by centrifugation. The cell pellet was resuspended in pre-warmed nucleofection solution, and 5 μg of plasmid DNA was added. The mixture was transferred to a cuvette and nucleofected. Immediately after nucleofection, the E8 medium supplemented with 10 μM Y-27632 was added to the cuvette, and the contents were transferred to the pre-coated 12-well plate. After 24 hours, the medium was replaced with E8 medium containing 0.4 μg/ml puromycin (Sigma-Aldrich) to select for transfected cells. The selection medium was removed after another 24 hours, and cells were maintained in standard E8 medium under routine culture conditions.
Surveyor nuclease assay
To assess the efficiency of sgRNA-mediated cleavage, genomic DNA was extracted from nucleofected BMD1 hiPSC line using the Genomic MINI kit (A&A Biotechnology), following the manufacturer’s instructions. The target region was amplified by PCR using KAPA Taq ReadyMix and specific primers (Supplementary Table 2). For heteroduplex formation, PCR products were mixed with Celery Juice Extract buffer and subjected to a thermocycling protocol consisting of denaturation at 95 °C for 5 min, cooling to 85 °C with a 30-sec hold, followed by gradual ramping to 25 °C at a rate of 5 °C per min, with an additional 30-sec hold at 25 °C. The reaction was then held at 4 °C to stabilize the heteroduplexes. The hybridized products were then incubated with the Celery Juice Extract (prepared according to the protocol described by Till et al. 17 containing Surveyor nuclease). The digestion reaction was carried out at 45 °C for 45 min. Digestion products were visualized by electrophoresis on a 2% agarose gel.
Construction of plasmid for the repair of the mutation in the DMD gene
The homology-directed repair (HDR) template was designed to contain left (5′) and right (3′) homology arms flanking the targeted deletion site. Silent mutation was introduced near the protospacer adjacent motif (PAM) to disrupt the PAM sequence and prevent Cas9-mediated re-cutting following successful repair. The HDR template was generated through high-fidelity PCR amplification using CloneAMP HiFi PCR Premix (Takara Bio) according to the manufacturer’s instructions. Primer sequences are listed in Supplementary Table 3. PCR products were analyzed on a 2% agarose gel; specific bands were excised and purified using the Zymoclean Gel DNA Recovery Kit (Zymo Research). Both the HDR template and the pcDNA3.1/Hygro (+) plasmid (Invitrogen #V87020) were digested with XhoI and MluI (NEB). The digestion reaction was carried out at 37 °C for 2 h. Digested DNA was separated on a 1% agarose gel, and the corresponding fragments were purified using Zymoclean Gel DNA Recovery Kit (Zymo Research) according to the manufacturer’s protocol.
For ligation, the digested pcDNA3.1/Hygro (+) vector and the HDR insert were combined with Quick Ligation Buffer and Quick Ligase (NEB). The mixture was incubated at 25 °C for 1 h. Transformation into E. coli and plasmid amplification were performed as described above. The correct insertion and sequence integrity of the repair template were confirmed by Sanger sequencing (Genomed S.A., Warsaw, Poland) using the primers listed in Supplementary Table 4. For genome editing in hiPSCs, nucleofection was performed (as described in section “Nucleofection”) using a combination of two plasmids: the HDR-containing pcDNA3.1/Hygro (+) vector and the pSpCas9(BB)-2A-Puro plasmid encoding Cas9 and the sgRNA.
Single cell cloning and genotyping of edited hiPSCs
Edited hiPSCs were seeded to obtain single-cell-derived clones. A total of 500 cells were transferred into each well of a 6-well plate. Once the colonies reached a visible size suitable for manual picking, individual clones were mechanically picked and transferred to separate wells of a 96-well plate. DNA was extracted from each hiPSC clone and subjected to PCR genotyping using KAPA2G Fast Genotyping Mix. The primers flanking the targeted upstream and downstream regions are listed in Supplementary Table 5.
Spontaneous differentiation of hiPSCs via embryoid bodies
HiPSCs (5,000 per well) were seeded into non-adherent U-shaped 96-well plates with Essential 6™ (E6) medium (Thermo Fisher Scientific) supplemented with 4 mg/ml polyvinyl alcohol (Sigma-Aldrich) and 10 μM Y-27632 and cultured for 5-7 days to form embryoid bodies (EBs). EBs were then transferred to Geltrex™-coated 48-well plates and maintained under spontaneous differentiation conditions for ∼14 days.
Karyotype analysis
Karyotyping was performed using the GTG-450 G-banding method (GTG-450, 15 mitoses/sample) by Kariogen cytogenic laboratory, Krakow, Poland.
Cardiac differentiation
Cardiac differentiation of hiPSCs was performed following established protocols,12,18,19 using small-molecule modulators of the Wnt/β-catenin pathway. CHIR99021, a GSK3β inhibitor, was applied to induce mesoderm formation, followed by Wnt pathway inhibition to promote cardiac mesoderm specification. HiPSCs (3 × 104) were seeded onto 24-well plates in E8 medium and cultured until reaching ∼90% confluency. On day 0, differentiation was initiated by treating cells with CHIR99021 (Sigma-Aldrich) for 24 h in RPMI-1640 medium (Sigma-Aldrich) supplemented with 2% B-27 minus insulin (RPMI/B27-ins; Thermo Fisher Scientific). After 24 h, the medium was replaced with fresh RPMI/B27-ins. On day 3, cells were treated with either XAV939 (MCE) for the BMD1 line or IWR-1 (Sigma-Aldrich) for the BMD2 line. From day 7, cultures were maintained in RPMI-1640 with B-27 containing insulin (RPMI/B27; Thermo Fisher Scientific). To enhance cardiomyocyte purity, metabolic selection was performed between days 13 and 19 in glucose-free RPMI medium (Gibco) supplemented with 4 mM sodium DL-lactate (Sigma-Aldrich). Cells were subsequently dissociated using the Multi Tissue Dissociation Kit 3 (Miltenyi Biotec), collected in RPMI-1640 with 20% fetal bovine serum (FBS; Biowest), centrifuged (200 × g, 5 min), and replated onto Geltrex™-coated wells in RPMI/B27 for further analysis. All experiments and analyses were performed on hiPSC-CMs between days 25 and 28 of differentiation.
Immunofluorescence staining
Immunofluorescence staining was performed on hiPSCs, EBs, hiPSC-CMs to detect cell type – specific markers. Cells were fixed with 4% paraformaldehyde (PFA, Santa Cruz Biotechnology) for 15 min at room temperature (rt), permeabilized with 0.1% Triton X-100 (BioShop Canada) in PBS (w/o Ca2+/Mg2+) for 15 min at rt, and blocked with 3% bovine serum albumin (BSA, BioShop Canada) in PBS (blocking buffer) for 1 h at rt. Primary antibodies, diluted in blocking buffer, were applied overnight 4°C (Supplementary Table 6). The following day, cells were washed with PBS and incubated with secondary antibodies (Supplementary Table 6) for 1 h at rt. Nuclei were counterstained with Hoechst 33342 (0.1 mg/ml). Imaging stained 2D cultures was performed using a Nikon Eclipse TS100 fluorescence microscope.
Reactive oxygen species level
The reactive oxygen species (ROS) levels in cardiomyocytes were assessed using the CellROX™ Deep Red Reagent (Thermo Fisher Scientific). Following the manufacturer’s instructions, 5 µM reagent was added to hiPSC-CMs and incubated at 37°C in the dark for 30 min. After incubation, the cells were rinsed with PBS and subsequently analyzed on an LSR Fortessa flow cytometer. Fluorescence intensity was quantified at the single-cell level as median fluorescence intensity (MFI), which limits the confounding effects of variable probe loading. To ensure consistent sample processing, all lines were seeded at identical densities and acquired under the same instrument settings.
Assessment of labile iron level
Labile iron pool (LIP) was assessed using the fluorescent probe Calcein-AM (Sigma-Aldrich). HiPSC-CMs were incubated with 5 μM Calcein-AM in PBS for 30 min at 37°C in the dark. After incubation, cells were washed with PBS, dissociated using the Multi Tissue Dissociation Kit 3 (Miltenyi Biotec), and centrifuged at 200 × g for 5 min. The pellet was resuspended in PBS containing 0.1 μg/ml Hoechst 33342. Fluorescence was measured using an LSR Fortessa flow cytometer and quantified at the single-cell level as median fluorescence intensity (MFI). Calcein fluorescence is quenched upon binding to labile iron; thus, increased MFI reflects decreased intracellular labile iron levels. MFI values were normalized within each isogenic pair to the mean MFI of the corresponding corrected control line (Ctrl1 or Ctrl2), which was set to 1.
Assessment of mitochondrial iron level
Mitochondrial iron levels were measured using Rhodamine B-[(1,10-phenanthroline-5-yl)-aminocarbonyl] benzyl ester (RPA; Squarix). HiPSC-CMs were incubated with 5 μM RPA in Hanks’ Balanced Salt Solution (HBSS; Thermo Fisher Scientific) for 15 min at 37 °C in the dark. Cells were then rinsed with HBSS, centrifuged at 200 × g for 5 min at 4 °C, and resuspended in fresh HBSS. After a second 15 min incubation under the same conditions, cells were centrifuged again and resuspended in HBSS containing 0.1 μg/ml Hoechst 33342. Fluorescence was measured at the single-cell level using an LSR Fortessa flow cytometer as MFI and normalized within each isogenic pair to the mean MFI of the corresponding corrected control line (Ctrl1 or Ctrl2), which was set to 1. RPA fluorescence is quenched upon binding to mitochondrial labile iron; therefore, increased MFI reflects lower mitochondrial iron levels. Normalized MFI values were used for comparisons between matched repaired and BMD hiPSC.
RNA – Based analysis
Total RNA was isolated using the Chomczynski and Sacchi method with Fenozol reagent (A&A Biotechnology) and chloroform. 20 RNA concentration and purity were assessed using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). 500-1000 ng of total RNA was used for reverse transcription (RT) performed with RevertAid reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer’s protocol. Gene expression was analyzed by quantitative real-time PCR (qRT-PCR) using SYBR Green JumpStart Taq Ready Mix (Sigma-Aldrich), as previously described. 21 Primer sequences are listed in Supplementary Table 7. qRT-PCR was performed on StepOnePlus thermocycler (Applied Biosystems) in StepOne v2.3 according to standard protocol. Expression levels were normalized to the reference gene EEF2. Data were analyzed using the ΔCt method.
Protein – Based analysis
For western blot analysis, hiPSC-CMs were lysed in Pierce RIPA Buffer (Thermo Fisher Scientific) with protease inhibitors, and total protein concentration was determined using the bicinchoninic acid (BCA) assay. A total of 20-50 μg protein was loaded onto 6–12% polyacrylamide gels, depending on target protein size. Gels were run at 80 V for 30 min and switched to 170 V for 1-2 h, followed by a wet transfer to a nitrocellulose membrane at 100 V at 4°C for 60-120 min. For protein detection, the nitrocellulose membrane was blocked in blocking buffer (5% w/v nonfat dry milk or BSA, 1× Tris-buffered saline, and 0.1% Tween 20) at rt for 1 h and incubated with primary antibody (Supplementary Table 8) at 4°C overnight, followed by incubation with HRP-conjugated secondary antibody at rt for 1 h (Supplementary Table 8). For protein size estimation, PageRuler™ Prestained Protein Ladder (10 to 180 kDa, Thermo Fisher Scientific) and HiMark™ Pre-stained Protein Standard (30 to 460 kDa, Thermo Fisher Scientific) were used. Signal was detected using Immobilon Western Chemiluminescent HRP Substrate (Sigma-Aldrich) and visualized using a ChemiDoc Imaging System (Bio-Rad). The intensities of the protein bands of interest were quantified by densitometric analysis using ImageJ software and normalized to GAPDH or ACTN2 as loading controls, with values expressed relative to control hiPSC-CM samples.
Statistical analysis
Numerical data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism. Due to substantial inter-line heterogeneity of hiPSC-derived cells, comparisons between control and dystrophic cell lines were performed within each patient using paired two-tailed Student’s t-test. Effects of H2O2 were analyzed using two-way ANOVA with cell line type (control vs dystrophic) and H2O2 treatment (untreated vs treated) as factors, followed by Sidak’s post hoc test to identify specific group differences. Each data point represents an independent biological differentiation. N refers to the number of independent hiPSC-CM lines, while n refers to the number of differentiations performed for each cell line. A p value < 0.05 was considered statistically significant (* p < 0.05, ** p < 0.01). Values were normalized to the pooled mean of the repaired line (Ctrl), calculated across two/three independent biological repeats.
Results
Strategies for CRISPR-Cas9 gene editing of DMD exon 45–47 and exon 3–9 deletions
This study focuses on two major DMD mutation hotspots, spanning exons 45-47 (BMD1) and 3-9 (BMD2), which are affected in the analyzed patient-derived hiPSC lines. To identify optimal CRISPR–Cas9 components for correcting DMD exon deletions, we assessed their ability to mediate HDR-based introduction of the wild-type sequence into patient-derived BMD hiPSCs, generating genetically corrected hiPSC lines (Figure 1(a)–(b)). Homology-directed repair (HDR) double-strand break (DSB) mechanism used for correction of DMD mutations in BMD patient-derived hiPSCs. The schematic illustrates the CRISPR/Cas9-mediated HDR strategy applied to repair two distinct DMD gene deletions: (a) Exons 45–47 (BMD1 patient) and (b) exons 3–9 (BMD2 patient). For each mutation, single-guide RNA (sgRNA) was designed to specifically bind sequences flanking the deletion site and direct Cas9 endonuclease to induce double-strand breaks (DSBs) at these locations. A donor repair template containing the missing DMD exons, flanked by left and right homology arms (LHA and RHA), was provided to guide precise repair via the HDR pathway. (c) Genomic DNA from BMD hiPSCs transfected with pSpCas9(BB)-2A-Puro encoding sgRNA was analyzed using Surveyor nuclease assay. Cleavage products, indicative of indel formation (left: 530 bp and 324 bp for BMD1 and right: 400 bp and 200 bp for BMD2), were detected in sgRNA1-treated cells but not in untreated control (CTRL). A positive control (POS) confirmed the assay’s functionality. The schematics illustrate the sgRNA target site and predicted Cas9 cleavage site within the amplified region. Ladder: DNA Marker 1 (100-1000 bp). (d) Generation of the HDR repair template. The template consisted of three fragments (two homological arms and the missing exons) to enable gene correction via HDR. Left: PCR-amplified DNA fragments. Right: final HDR template generated by overlap extension PCR (OE-PCR). Ladders: DNA Marker 1 (100-1000 bp) and IDEAL2 DNA Ladder (700–9276 bp). E. PCR-based genotyping of DMD repaired clones. For BMD1, one successfully corrected clone was identified, showing a specific PCR amplicon of 1439 bp spanning exons 45-48. For BMD2, three successfully corrected clones were obtained, each showing a specific PCR amplicon of 1479 bp spanning exons 3-9. Ladder: IDEAL2 DNA Ladder (700–9276 bp). Gel images were cropped, and lanes were rearranged for clarity; therefore, samples are not shown in the original order. Uncropped raw gel images were provided to the journal.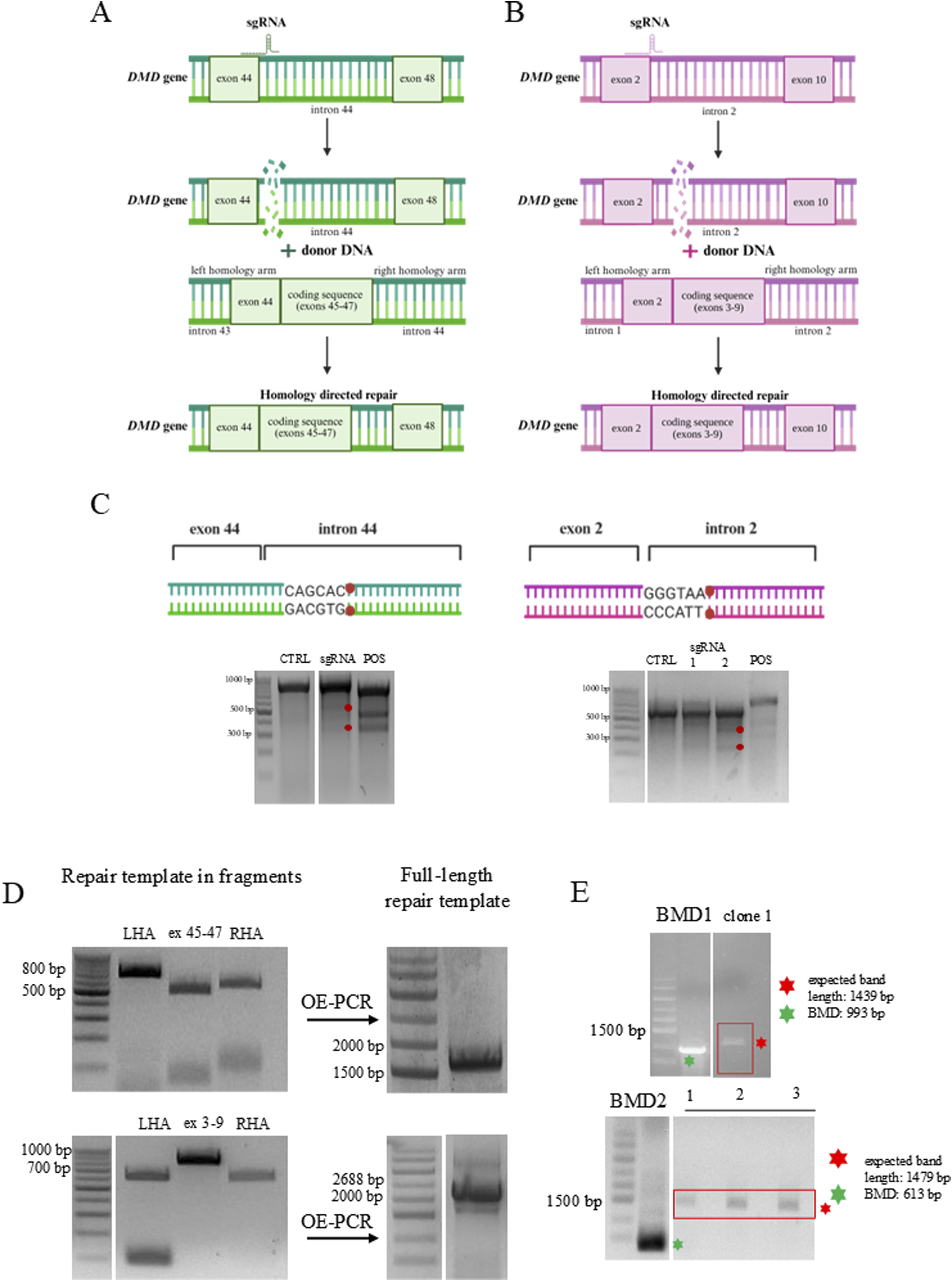
To optimize the genome editing strategy, we designed sgRNAs targeting intronic regions flanking the deletion breakpoints, aiming to restore the full-length dystrophin coding sequence via CRISPR-Cas9. In vitro screening using the Surveyor assay identified the most efficient sgRNA candidates, paired with SpCas9 from Streptococcus pyogenes. In BMD hiPSCs, two distinct cleavage bands (BMD1: 530 bp and 324 bp; BMD2: 400 bp and 200 bp) confirmed efficient Cas9-mediated cleavage (Figure 1(c)).
To facilitate precise genome correction, a donor DNA template for HDR was designed, containing overlapping homology arms flanking the deletion site (Figure 1(a)–(d)). This HDR template was co-delivered with plasmids encoding the selected sgRNAs and SpCas9 into BMD hiPSCs via nucleofection. Following the expansion of single-cell clones, successful genome editing events were validated by genomic PCR (Figure 1(e)). The corrected lines were designated as Ctrl1 and Ctrl2.
Karyotype analysis confirmed a normal diploid chromosomal complement both prior to and following CRISPR/Cas9-mediated correction of the mutations (Figure 2(a)). In addition, the lines exhibited expression of pluripotency markers, including OCT3/4, and SSEA4 and TRA-1-60 (Figure 2(b)). Upon spontaneous differentiation through EBs formation, cells expressed markers representative of all three germ layers: vimentin (mesoderm), GATA4 (endoderm/mesoderm) and neurofilament heavy chain (NFH; ectoderm) (Figure 2(c)), described previously for BMD2.
8
Characterization of corrected and BMD1 hiPSC lines. (a). Karyotypic analysis of Ctrl1 and BMD1 hiPSC lines. (b) Immunocytochemistry staining for pluripotency markers NANOG, OCT-3/4 and SSEA4. Scale bar: 100 μm. (c) Immunofluorescent staining for endo- (GATA4), ecto- (neurofilament heavy chain – NH) and mesoderm (GATA4, vimentin) markers after spontaneous differentiation. Scale bar: 100 μm. Full characterization of the generated hiPSC line was reported previously.
8

Restoration of full-length dystrophin expression in hiPSC-CMs
In the next step, the repaired and their parental BMD lines were differentiated into hiPSC-CMs as outlined in the schematic (Figure 3(a)). CHIR99021 was used to initiate differentiation. For WNT/β-catenin pathway inhibition, IWR-1 was used for Ctrl2 and BMD2, while XAV-939, a tankyrase inhibitor, was applied to Ctrl1 and BMD1, due to reduced differentiation efficiency. Notably, both inhibitors act at the level of WNT/β-catenin pathway suppression, and no differences in cardiac identity were observed between lines differentiated with IWR-1 or XAV-939. At the mRNA level, we confirmed the presence of the transcript containing the missing exons in both repaired lines, confirming the successful integration of the missing exons (Figure 3(b)). Characterization of hiPSC-CMs differentiation and restoration of dystrophin expression by CRISPR-mediated gene correction. (a) Schematic timeline of differentiation from hiPSCs to hiPSC-CMs. (b) Successful knock-in was confirmed by specific RT-PCR amplicons spanning exons 45-48 in BMD1 and exons 3-9 in BMD2. Ladder: DNA Marker 1 (100-1000 bp). Gel images were cropped, and lanes were rearranged for clarity; therefore, samples are not shown in the original order. Uncropped raw gel images were provided to the journal. (c) Representative images showing expression of dystrophin (DMD) and cardiac troponin T (cTnT) in repaired and BMD hiPSC-CMs. Scale bar: 100 μm. (d) Representative western blot analysis of the full-length dystrophin isoform Dp427 in repaired (Ctrl) and BMD hiPSC-CMs. α-Actinin (ACTN2) was used as a loading control.
Repaired and BMD hiPSC-CMs showed expression of dystrophin (DMD) and cardiac markers, cardiac troponin T (cTnT), and α-sarcomeric actinin (ACTN2), confirming their cardiac identity and dystrophin restoration (Figure 3(c)–(d)). Western blot analysis showed that the deletions of exons 45–47 (BMD1) and 3–9 (BMD2) do not significantly alter dystrophin molecular weight, and CRISPR-mediated repair did not introduce any unintended modifications affecting expression or stability.
Disturbed iron levels and impaired iron regulatory network with reduced mitoNEET level in BMD hiPSC-CMs
In BMD hiPSC-CMs, basal levels of ROS were elevated compared to repaired cells (Figure 4(a)). Upon treatment with 50 µM H2O2, ROS production further increased, indicating heightened oxidative stress susceptibility in BMD cells (Figure 4(a)). While these effects did not reach statistical significance, the increase in ROS in two separate lines of BMD cardiomyocytes was evident. Such trends also apply to other changes, as demonstrated below. Disturbances in iron homeostasis in BMD hiPSC-CMs. A. Level of oxidative stress in control (Ctrl) and BMD hiPSC-CMs in basal conditions and in response to 50 µM H2O2 treatment. Data are presented as median fluorescence intensity normalized to corrected cells (ctrl = 1); N=2, n=3. (b and c). Cytosolic (B) and mitochondrial (C) iron levels assessed by flow cytometry using Calcein-AM (B) and RPA (C). Values represent median fluorescence intensity normalized to corrected cells (ctrl = 1); N=2, n=3. (d) Representative western blot showing mitoNEET protein expression in Ctrl and BMD hiPSC-CMs. Reduced mitoNEET levels in BMD cells were restored following DMD gene correction. Densitometric quantification of mitoNEET protein expression; N = 2, n = 3–4. (e) Gene expression of key iron-regulatory genes (CISD1, FTH1, SLC40A1, TF, and TFRC) in BMD hiPSC-CMs. Values represent relative expression normalized to corrected cells (ctrl = 1); N=2, n=2-3. N = line replicates, n = differentiation replicates. Statistical analysis: Two-way ANOVA (A), paired two-tailed Student’s t-test (B-E). * p < 0.05, ** p < 0.01, *** p < 0.001.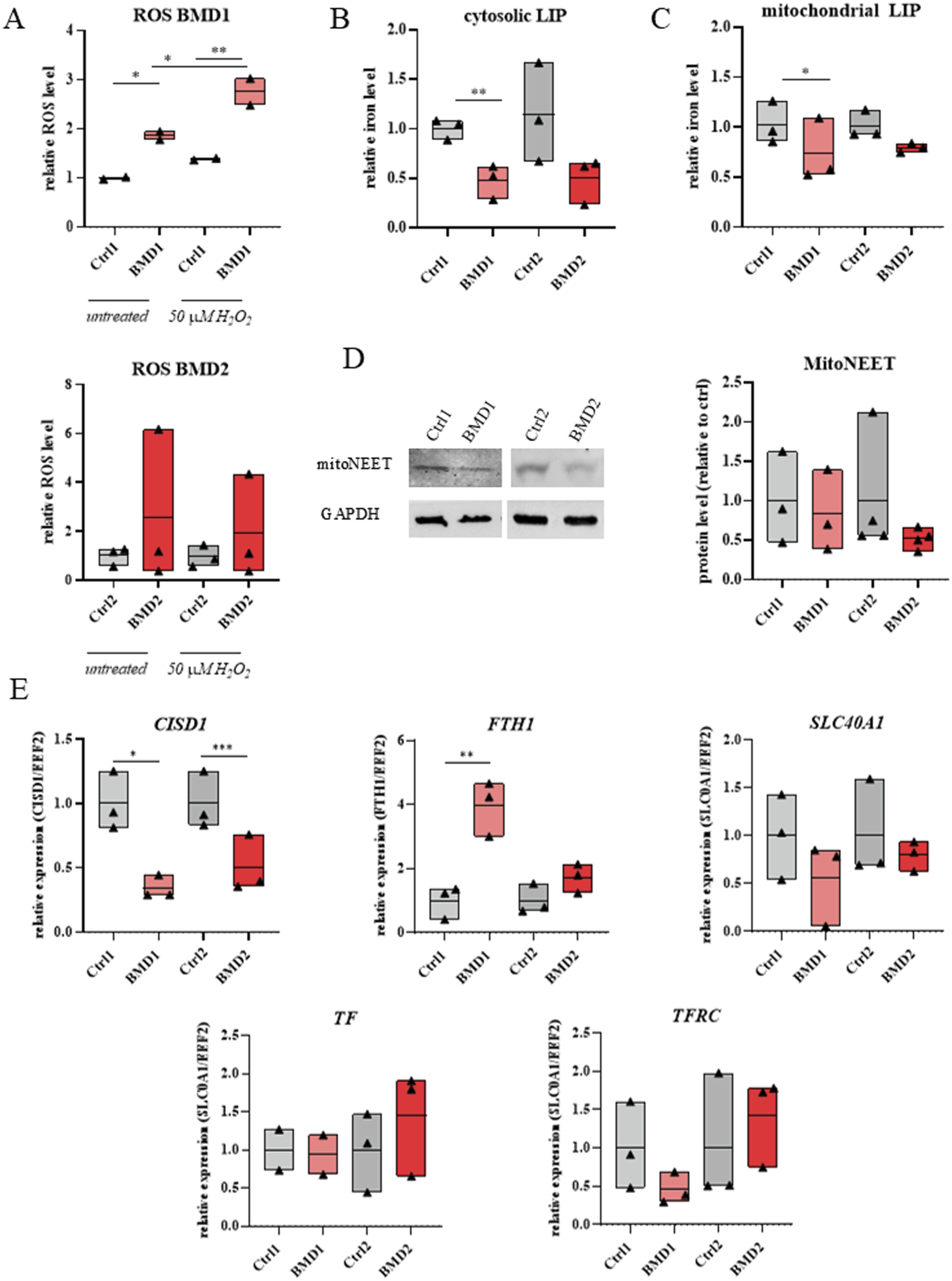
BMD hiPSC-CMs exhibited reduced cytoplasmic free iron levels relative to their repaired genome-corrected counterparts (Figure 4(b)). Mitochondrial labile iron was also decreased, although to a lesser extent than in the cytoplasm (Figure 4(c)). Western blot analysis of BMD1, BMD2, and repaired hiPSC-CMs revealed a trend toward reduced levels of the mitochondrial protein mitoNEET in the BMD lines, as illustrated by representative blots and densitometric quantification (Figure 4(d)).
Gene expression analysis of iron homeostasis–related markers revealed distinct alterations in BMD hiPSC-CMs. In both BMD lines, CISD1, which encodes mitoNEET, was downregulated in comparison to respective control lines (Figure 4(e)). In contrast, expression of FTH1, which produces the ferritin heavy chain subunit responsible for intracellular iron storage, was markedly increased in BMD1 (vs. Ctrl1) and showed a trend toward upregulation in BMD2 (vs. Ctrl2) (Figure 4(e)). The iron exporter SLC40A1, a gene for ferroportin, tended to be downregulated in BMD (Figure 4(e)). No significant changes were detected in the level of TF and TFRC, encoding transferrin and transferrin receptor, respectively (Figure 4(e)).
Discussion
In this study, we established and characterized BMD patient-specific hiPSC lines and restored full-length dystrophin expression via CRISPR/Cas9-mediated genome editing. Upon directed differentiation into cardiomyocytes, both repaired and BMD hiPSC lines provided a relevant human model to investigate the cardiac manifestations of BMD and to explore the impact of BMD-associated mutations on iron homeostasis.
We observed that increased ROS levels in BMD hiPSC-CMs were accompanied by upregulation of FTH1 and a concomitant reduction in both cytoplasmic and mitochondrial LIPs. 22 This pattern is consistent with a canonical oxidative stress response in which FTH1 expression is induced to enhance iron sequestration and limit the availability of redox-active Fe2+. By reducing the LIP, ferritin-mediated buffering restricts Fenton reactions and thereby constrains further ROS production.23,24 Thus, despite elevated oxidative stress, BMD hiPSC-CMs exhibit a reduction rather than expansion of the redox-active iron pool, suggesting activation of a compensatory iron-buffering program. Importantly, these findings extend our previous observations in DMD hiPSC-CMs 12 and suggest that iron homeostasis is disrupted across dystrophinopathies in a severity-dependent manner. Whereas DMD hiPSC-CMs exhibited an expansion of the labile iron pool, BMD cells showed a reduction consistent with enhanced ferritin-mediated iron sequestration. Whether these differences in labile iron pool levels between BMD and DMD cardiomyocytes reflect various kinetics dependent on the type of DMD mutation requires further investigation. Importantly, in both contexts, CRISPR/Cas9-mediated correction of DMD mutations restored iron-related alterations in patient-derived cardiomyocytes.
To further explore the molecular signatures associated with altered iron metabolism in BMD hiPSC-CMs, we analyzed the expression of key genes involved in iron regulation. The expression of CISD1 was reduced in BMD hiPSC-CMs and confirmed by a marked decrease in mitoNEET protein level. This agrees with the lower mitoNEET levels in DMD hiPSC-CMs 12 and indicates that regulation of CISD1 expression occurs both at the mRNA and protein level. MitoNEET is a CDGSH iron-sulfur domain-containing protein that coordinates a [2Fe-2S] cluster. It is localized to the outer mitochondrial membrane, where it plays a key role in binding iron via Fe-S clusters, as well as in regulating energy metabolism. Interestingly, among all murine tissues, CISD1 exhibits the highest expression in the heart. 13 Mice with CISD1 deletion develop heart failure and show elevated ROS levels. Additionally, a significantly elevated iron content was detected in mitochondria isolated from cardiac muscle of CISD1-deficient mice compared with control mice. 14 Although our data indicate a reduced LIP, these findings suggest that altered iron compartmentalization and mitochondrial redox regulation – rather than simple iron overload – may contribute to oxidative stress in BMD hiPSC-CMs. Importantly, this evidence remains correlative, as no perturbation experiments were performed to establish a causal role of CISD1/mitoNEET in the observed phenotype.
Notably, BMD hiPSC-CMs also exhibited a tendency toward downregulation of SLC40A1, encoding the iron exporter ferroportin, and TFRC, encoding the transferrin receptor. Although these changes did not reach statistical significance, the reproducible trend suggests altered regulation of iron import and export in BMD hiPSC-CMs, potentially contributing to disturbing iron homeostasis.
While hiPSC-CMs represent a relevant human cardiac model, their developmental immaturity must be considered when interpreting iron-related phenotypes, as metabolic and mitochondrial deficits may manifest differently compared to adult myocardium. 25 They display differences in electrophysiological properties, metabolic profiles, and structural organization, which may affect the interpretation of experimental results, particularly in the context of late-onset cardiac diseases. 26
Together, these findings suggest that iron dysregulation is a shared feature of dystrophinopathies yet manifests differently depending on disease severity. In contrast to the iron-overload phenotype observed in DMD, BMD hiPSC-CMs exhibit a ferritin-associated iron-withholding profile despite elevated ROS.
Limitations of the study
The present study has several limitations that merit consideration. While we observe consistent changes in iron homeostasis – related pathways in BMD hiPSC-CMs, we did not perform direct functional assessments of mitochondrial or cellular bioenergetics. Moreover, the Calcein-AM and RPA flow-cytometry readouts were quantified as normalized MFI signals, and additional validation controls (e.g., iron chelator displacement assays) would further strengthen assay specificity and help confirm signal directionality. As a result, the relationship between iron dysregulation and cardiomyocyte energetic function cannot be fully delineated. Second, we have used hiPSC-CMs which represent a relevant human cardiac model, however, their developmental immaturity must be considered when interpreting iron-related phenotypes, as metabolic and mitochondrial deficits may manifest differently compared to adult myocardium. They display differences in electrophysiological properties, metabolic profiles, and structural organization, which may affect the interpretation of experimental results, particularly in the context of late-onset cardiac diseases. 13 Our analyses were performed at a defined differentiation stage and therefore do not address the temporal dynamics of iron dysregulation during cardiomyocyte differentiation or maturation. Longitudinal studies assessing multiple developmental or maturation stages would be valuable for clarifying the timing and progression of these changes.
A limitation of our study is that we investigated only two isogenic pairs of BMD hiPSC-CMs, which may explain the apparent trend in changes, although it is not yet statistically significant. Although two to three independent differentiations were performed for each line, further studies using cardiomyocytes derived from a larger number of patients with this rare disease will be necessary to validate these results. In addition, our findings were not validated in patient cardiac tissue or in vivo models, and therefore their physiological relevance at the tissue and organismal level requires further investigation. Finally, although we have previously showed iron homeostasis dysregulation in DMD hiPSC-CMs, 12 a direct comparative analysis between BMD and DMD cells was not performed. However, based on the available data, a tendency toward reduced mitoNEET levels was observed in both BMD (this study) and DMD 12 hiPSC-CMs, with an average decrease of approximately 19% compared to control conditions. In DMD hiPSC-CMs, iron accumulation was statistically significant, whereas in BMD cells, we observed reduced LIPs. Increased ROS levels were detected in both models, although statistical significance was not consistently achieved across all lines. These observations should be considered preliminary, and a direct side-by-side analysis of more BMD and DMD hiPSC-CMs would be required to draw definitive conclusions regarding the relationship between iron imbalance, mitoNEET levels, and disease severity. Despite listed limitations, the study provides a systematic characterization of iron-related molecular changes in diseased cardiomyocytes and establishes a foundation for future mechanistic and functional investigations. We show that even in the presence of truncated dystrophin, BMD hiPSC-CMs already exhibit marked disturbances in iron homeostasis, as evidenced by attenuated expression of the mitochondrial iron regulator mitoNEET. Our results suggest that restoration of full-length dystrophin expression may contribute to maintaining iron balance. However, additional studies are needed to fully elucidate the mechanisms.
Supplemental material
Supplemental material - Gene editing restores full-length dystrophin and affects iron homeostasis in hiPSC-derived cardiomyocytes from becker muscular dystrophy patients
Supplemental material for Gene editing restores full-length dystrophin and affects iron homeostasis in hiPSC-derived cardiomyocytes from Becker Muscular Dystrophy patients by Marta Przymuszała, Joanna Kwiatkowska, Jarosław Meyer-Szary, Karolina Śledzińska, Jolanta Wierzba, Agnieszka Łoboda, Jacek Stępniewski, Urszula Florczyk-Soluch and Józef Dulak in Journal of Neuromuscular Diseases.
Footnotes
Acknowledgments
Schematic Figure 1(a)–(c) and ![]() were created with BioRender.com.
were created with BioRender.com.
Ethical considerations
Ethical approval for the study was obtained from Gdańsk Medical University (Approval No. NKBBN/402/2020) and the research was carried out in accordance with the declaration of Helsinki.
Consent to participate
Patients signed an informed consent for participation and sample collection.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the MAESTRO grant (2018/30/A/NZ3/00412) from the National Science Centre to J.D.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the authors utilized AI tools to enhance the language. After using these tools, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.