
Abstract
Muscle fibrosis is a key pathological feature of Duchenne muscular dystrophy (DMD) and is closely associated with disease progression. Fibroadipogenic progenitors (FAPs) are major contributors to fibrosis, yet the precise mechanisms remain unclear. To investigate FAP dynamics and lineage specification, we generated dual-reporter mice (PRURD2) by crossing D2.B10-Dmdmdx/J (D2-mdx) mice with FAP and brown/beige adipose tissue (BAT) reporter lines. Corresponding control mice (PRURDBA) were established on the DBA/2J background. At 12 months, heart, diaphragm, and tibialis anterior (TA) muscles were collected for histological analysis. FAPs were isolated via FACS and subjected to single-cell RNA sequencing. PRURD2 mice exhibited increased fibrosis across all muscles compared to controls (p < 0.01) and a significant rise in PDGFRα-GFP + FAPs (p < 0.05). UMAP clustering identified 11 distinct FAP subpopulations, with the fibrosis-associated CD55 cluster enriched in PRURD2 mice. Pseudotime analysis showed lineage progression from progenitor clusters toward the fibrogenic CD55 cluster. CellChat analysis indicated increased interactions in PRURD2 mice involving fibrosis-related pathways like COLLAGEN, TGF-β, WNT, NOTCH, and ANGPTL. Additionally, fibrosis-related signaling pathways such as THY1, TWEAK, EPHA, EPHB, and SEMA6 showed increased interactions among FAP clusters in PRURD2 mice. Differential gene expression analysis revealed top upregulated genes including Cxcl13, Cxcl3, Ly6d, Klk1, Fgf23, Serpinb2, Mmp13, Ccl17, Postn, and Adam12. PRURD2 mice develop severe fibrosis in skeletal and cardiac muscle, driven by FAP-induced signaling pathways and genes. This model is valuable for understanding muscle fibrosis in DMD and developing anti-fibrotic therapies.
Keywords
Introduction
Duchenne muscular dystrophy (DMD) is a genetic neuromuscular disorder affecting approximately 1 in 3500–6000 live-born males. It is characterized by progressive muscle degeneration and weakness caused by mutations in the dystrophin gene. 1 The absence of functional dystrophin compromises the structural integrity of the sarcolemma, rendering myofibers susceptible to contraction-induced injury. This results in chronic muscle degeneration and the progressive replacement of functional contractile material with fibrotic and adipose tissue in both cardiac and skeletal muscles. 2 The accumulation of fibrosis reduces the amount of healthy muscle fibers, leading to a marked decrease in contractile function. As a result, anti-fibrotic approaches have gained increased interest as promising therapeutic strategies for ameliorating disease progression in DMD. 3 Notably, fibrosis is among the earliest pathological features in the dystrophic heart, making its first appearance in DMD patients before 10 years of age. 4 Cardiac fibrosis contributes to heart failure and arrythmia by replacing functional myocardium, increasing workload demand on the remaining cardiomyocytes, and disrupting the tracts of electrical conduction pathways. 5 Although corticosteroids offer some therapeutic benefits, there remains a need for more effective treatments that directly target the underlying causes of fibrosis. 6
Recently, mesenchymal stromal cells, including Fibroadipogenic progenitors (FAPs), have garnered significant attention as key contributors to fibrosis in Duchenne muscular dystrophy (DMD). These FAPs, first described by Uezumi et al. 7 and Joe et al. 8 in 2010, reside in muscle interstitium, proliferate in response to muscle injury, and are characterized by the expression of platelet-derived growth factor receptor alpha (PDGFRα) (commonly used as a specific molecular marker for screening FAPs). Under normal conditions, FAPs play a critical role in maintaining healthy skeletal muscle homeostasis. However, under chronic injury or disease conditions, their excessive proliferation and pathological adipogenesis and fibrosis lead to fibrotic and fatty degeneration of muscle tissue. Several single-cell transcriptomic studies have reported the expression of fibrosis-related genes in FAPs.9,10 Nevertheless, these investigations rely on the DMD mouse model (C57BL/10ScSn-Dmdmdx/J), which exhibits only mild fibrosis and fails to recapitulate the extensive fibrosis observed in human DMD accurately. Moreover, while most research has focused on skeletal muscle FAPs, their role in DMD-related cardiac fibrosis, arrythmia, and contractile dysfunction remains poorly understood. 11
We recently discovered that β3 adrenergic receptor (B3AR) agonists can drive FAPs toward a brown/beige adipose tissue (BAT) phenotype, supporting muscle regeneration without fibrotic differentiation.12,13 This BAT differentiation of FAPs is marked by high mitochondrial content and expression of uncoupling protein 1 (UCP1). PDGFRα and UCP1 double-reporter mice allow in vivo tracking of FAP-BAT differentiation in response to various muscle injuries. In this study, we developed and characterized a FAP-BAT double-reporter mouse model on the D2.B10-Dmdmdx/J (D2-mdx) background (termed PRURD2 mice), a strain known to exhibit severe cardiac and skeletal muscle fibrosis comparable to that observed in DMD patients.14,15 Using this model, we monitored FAP lineage differentiation and fibrosis in both cardiac and skeletal muscle types. Results from this study provide novel insights into the in situ differentiation of FAPs in both cardiac and skeletal muscle and offer a deeper understanding of the cellular mechanisms driving muscle fibrosis in DMD.
Methods
Animals and experimental design
All procedures and handling of the animals were approved by the Institutional Animal Care and Use Committee at the San Francisco VA Medical Center. To generate FAP-BAT double reporter DMD mouse line (PR/UR), we crossed PDGFRα-GFP (hallmark of FAP, JAX #007669, B6.129S4-Pdgfra < tm11(EGFP)>Sor/J) 16 and UCP1-RFP (hallmark of BAT, JAX #026690, STOCK Tg(Ucp1-luc2,-tdTomato)1Kajim/J) 17 reporter mice. These mice were further backcrossed onto the D2-mdx or D2-wild type (DBA/2J) mice for six generations to generate PDGFRα-GFP/UCP1-RFP/D2-mdx (PRURD2) or PDGFRα-GFP/UCP1-RFP/DBA/2J (PRURDBA) strains, respectively. Mice were housed under standard conditions (12 h light/dark cycle, with free access to food and water). A total of nine male PRURDBA and nine male PRURD2 mice were used in this study. At twelve months of age, animals were sacrificed, and their heart, diaphragm, and tibialis anterior muscles (TA) were collected for analysis. Five of the nine mice underwent body weight and forelimb grip force assessments using a commercial dynamometer (BIO-GRIPGS, Bioseb) prior to euthanasia. A schematic overview of the experimental workflow is presented in Figure 1(a).
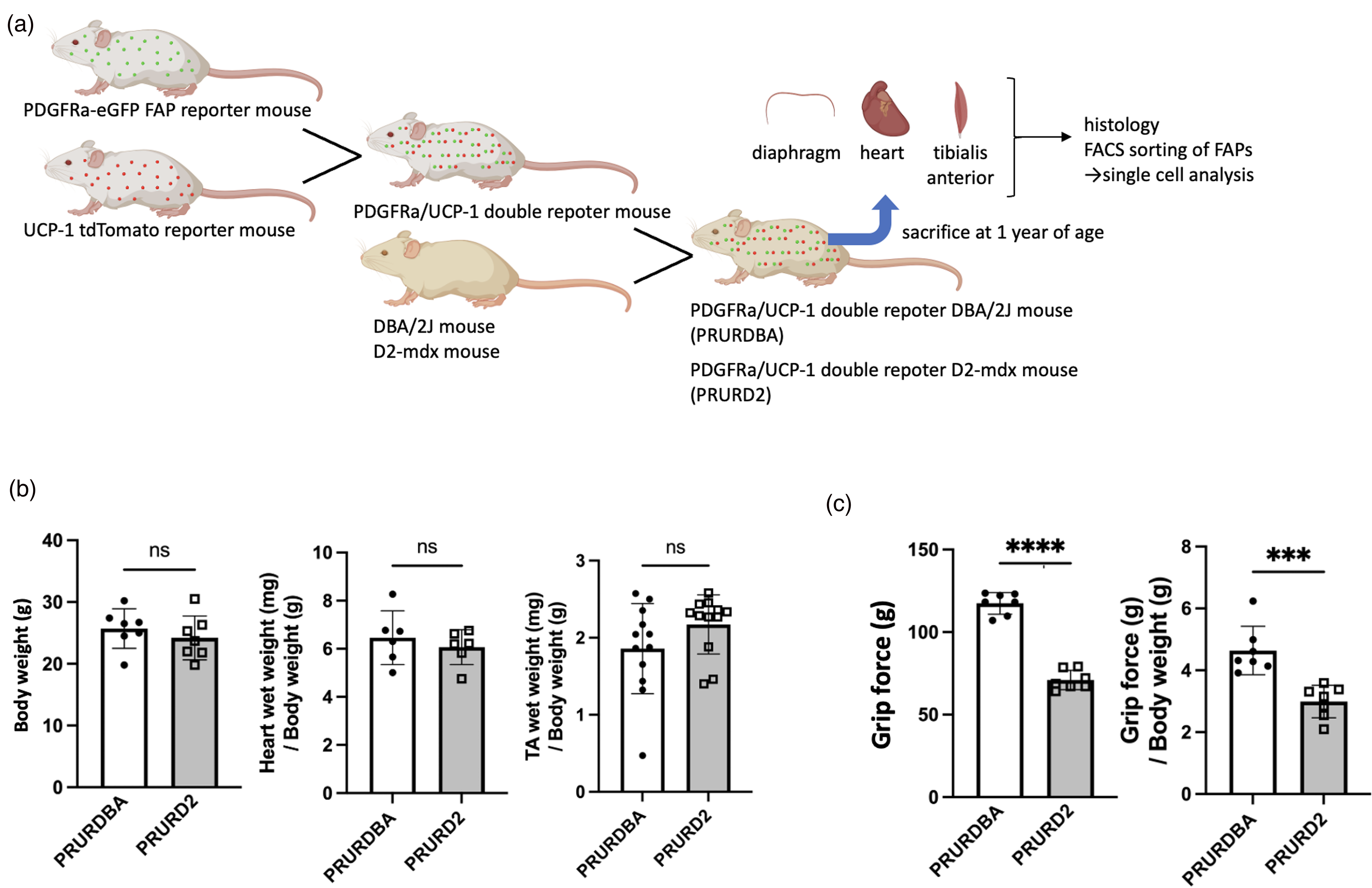
Experimental protocols and physiological assessment of PRURD2 and PRURDBA mice. (a) Schematic overview of the generation of FAB-BAT double reporter mice and the experimental design. (b and c) Comparison of body weight and muscle wet weights of the heart and tibialis anterior (TA), normalized to body weight (b), and grip force and grip force normalized to body weight (c) between PRURDBA (FAP-BAT double reporter DBA/2J) mice and PRURD2 (FAP-BAT double reporter D2) mice at 1 year of age. Data are presented as mean ± SD with individual values. ***p < 0.001; ****p < 0.0001 comparisons between indicated groups.
Muscle harvesting and histology
PRURDBA (n = 6) and PRURD2 (n = 6) mice were heparinized, and the heart was perfused with 10 mL of PBS, followed by 20 mL of 4% paraformaldehyde (PFA)- phosphate-buffered saline (PBS) through a 23-gauge needle. The heart, diaphragms, and TA were excised and post-fixed in 30% sucrose overnight at 4 °C for cryoprotection and then embedded in Optimal Cutting Temperature (OCT) compound (Tissue-Tek). 18 Prior to freezing, the wet weights of the heart and TA muscles were measured. Cryosections (10 μm) were prepared for histological staining to assess fibrosis (Masson Trichrome; StatLab), calcification (Alizarin Red; LabChem), 19 or immunofluorescence to evaluate fibrosis markers, such as alpha-smooth muscle actin (α-SMA). For immunofluorescence, sections were fixed with 4% paraformaldehyde for 10 min and washed in 0.1% Tween in PBS for 5 min three times. For detecting the unique PDGFRα-GFP and UCP1-RFP signals in the reporter mice, sections were mounted with anti-fading mounting media (Antifade Mounting Medium with DAPI, H-1800, Vector Labs). Sections were blocked with 1% bovine serum albumin (BSA) in PBS for 1 h at room temperature and then incubated with primary antibodies against laminin (1:200; made in rat; Invitrogen Cat No. MA1-06100) and anti-α-SMA (1:500; made in Goat; Sigma Aldrich Cat No. SAB2500963) overnight at 4°C. Sections were washed with 0.1% Tween in PBS 3 times for 5 min each wash and then incubated with donkey anti-rat Alexa Fluor 405 (1:200; Abcam Cat No. ab175670) and donkey anti-goat Alexa Fluor 647 (1:200; Invitrogen Cat No. A21447) in 1% BSA. After washing 3 times with PBS, slides were mounted with Fluoromount-G (Invitrogen). Stained sections were then visualized using the all-in-one fluorescent microscope BZ-X810 (KEYENCE). The fibrotic area was quantified as a percentage of total muscle area using BZ-X analyzer software (KEYENCE).
Isolation of fibroadipogenic progenitor cells
Heart, diaphragm, and bilateral TA muscle were harvested from the PRURDBA mice (n = 3) and PRURD2 mice (n = 3). Using sterile micro-dissecting scissors, excess adipose tissue, tendons, and connective tissue were removed. Tissues were finely minced and subjected to enzymatic digestion in a sterile biosafety cabinet. The digestion media consisted of Collagenase Type III (Worthington Biochemical Corporation, Cat# NC0021929) at a concentration of 15 mg per gram of tissue, prepared in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, Cat# 11965084) supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat# 10437028) and 1% penicillin/streptomycin (P/S) (Gibco, Cat# 15140122). Tissue samples were incubated in this digestion media at 37 °C for 70 min, with mechanical trituration using an 18-gauge needle performed midway through the incubation to facilitate dissociation. 20 Samples were washed with PBS and subjected to a second enzymatic digestion using 0.25% trypsin-EDTA (Gibco, Cat# 25200056) at 37 °C for 12 min. Digestion was quenched with FBS, and the resulting cell suspension was passed through a 40 μm nylon mesh filter. Cells were then washed in PBS containing 2% FBS, and erythrocytes were lysed using ammonium-chloride-potassium (ACK) lysing buffer (Quality Biological, Cat# 188-156-101) for 5–7 min on ice. Samples were then washed with 2% FBS in PBS and resuspended in 100 µL flow cytometry buffer, composed of PBS with 2% FBS, 5 mM EDTA (Corning, #MT-46034CI), and 2 mM HEPES (Quality Biological, #118-089-721) per 1 g of starting tissue weight. FAPs were isolated by fluorescence-activated cell sorting (FACS) based on PDGFRα-GFP reporter expression, enabling enrichment of the FAP population.
Single-cell RNA sequencing and data analysis
Single-cell RNA sequencing (scRNAseq) was performed on FACS-isolated FAPs from the diaphragm, heart, and TA of PRURDBA mice (n = 3) and PRURD2 mice (n = 3). For each tissue, FAPs from three mice of the same genotype were pooled and loaded into a single lane of a 10x Genomics Chromium controller, resulting in six separate scRNAseq libraries (one per tissue per genotype). Libraries were prepared using the 10x Genomics Chromium Single Cell 3′ Reagent kit (10x Genomics, NEXT Gel Beads-in-emulsion (GEM) Single Cell 3′ version 3.1, dual index PN-1000268), according to the manufacturer's protocol for GEM generation, cDNA production, and library construction. Sequencing of constructed libraries was carried out on Illumina NovaSeq.
Raw sequencing reads were processed using CellRanger (version 9.0.2) to demultiplex, align reads to the mm10 mouse reference genome (provided by 10x Genomics), and quantify unique molecular identifiers (UMIs). Doublets were removed using DoubletFinder based on input cell concentrations and expected multiplet rate estimates as recommended by 10x Genomics. Downstream analysis was performed using Seurat (v5.0). For each tissue, dataset was filtered, clustered, and visualized using uniform manifold approximation and projection (UMAP). Datasets were then integrated using canonical correlation analysis (CCA) integration, following the Seurat integration tutorial. Raw, unintegrated gene counts were pseudo-bulked by experimental group, and differential expression analysis was performed using NOISEQ.21,22 Pseudotime trajectory analysis was carried out using Monocle3 and cell-cell communication between each FAP subpopulation was performed using CellChat (version 1.1.3).
Statistical analysis
Prism software (Version 7.0a; GraphPad Software) was used for statistical analysis. Unpaired two-tailed t-tests were used to assess differences between experimental groups. Data are presented as mean ± SD with individual values shown where applicable. Statistical significance was defined as p < .05.
Results
FAP-BAT double reporter D2-mdx mice show severe fibrosis and calcification in both cardiac and skeletal muscle
The body weights and wet weights of the heart and TA muscles normalized by the body weight were comparable between PRURD2 and PRURDBA mice (body weight: 25.7 ± 3.2 g vs. 24.2 ± 3.5 g, p = 0.414; heart weight/body weight: 6.5 ± 1.1 mg/g vs. 6.1 ± 0.7 mg/g, p = 0.488; TA weight/body weight: 1.9 ± 0.6 mg/g vs. 2.2 ± 0.4 mg/g, p = 0.136) (Figure 1(b)). In contrast, forelimb grip force and grip force normalized by body weight were significantly reduced in PRURD2 mice compared to PRURDBA (grip force: 117.4 ± 6.6 vs 70.9 ± 5.9 g, p < 0.0001; normalized grip force: 4.6 ± 0.8 g/g vs 3.0 ± 0.5 g/g, p < 0.001) (Figure 1(c)). Histological analysis revealed diffusely distributed fibrotic areas in the diaphragm and TA of PRURD2 mice, while cardiac fibrosis was predominantly focal and localized to the periphery of the right ventricle (Figure 2(a)). Quantitative analysis confirmed significantly increased fibrotic areas in PRURD2 mice compared to PRURDBA mice across all tissues (Heart: 2.2 ± 0.9 vs 7.6 ± 2.6%, p < 0.001; Diaphragm: 4.7 ± 2.7 vs 30.6 ± 14.7%, p < 0.01; TA: 3.8 ± 1.9 vs 16.3 ± 7.4%, p < 0.01) (Figure 2(b)). Intramuscular calcification was observed only in tissues from PRURD2 mice (Figure 2(c)).
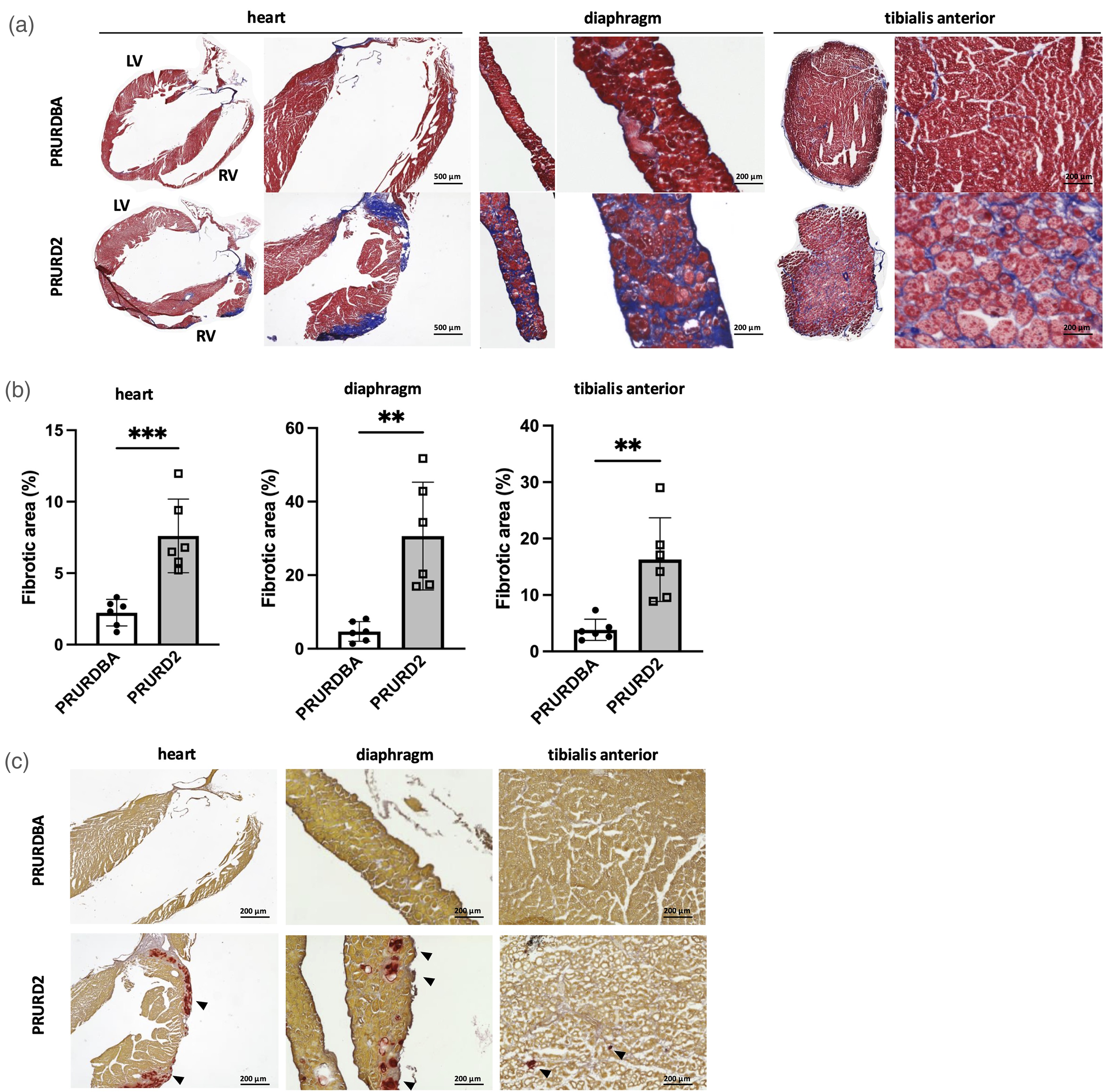
Histological evaluation of cardiac and skeletal muscle fibrosis and calcification in FAP-BAT double reporter D2-mdx (PRURD2) and DBA/2J (PRURDBA) mice. (a) Representative Masson's Trichrome stained section of the heart, diaphragm, and tibialis anterior (TA) muscles. Cardiac sections are labeled to indicate the right ventricle (RV) and left ventricle (LV). (b) Quantification of fibrotic area as a percentage of total muscle area in each tissue. (c) Representative Alizarin Red-stained sections of the heart, diaphragm, and TA muscles, showing areas of intramuscular calcification (black arrowheads) observed in PRURD2 but not PRURDBA mice. Data are presented as mean ± SD with individual values. **p < 0.01 and ***p < 0.001 comparisons between indicated groups.
Increased FAP and UCP1 reporter signal in fibrotic areas of PRURD2 mice
PRURD2 mice had a significant increase in PDGFRα-GFP positive cells compared to PRURDBA mice in the heart, diaphragm, and TA muscles (Heart: 5.2 ± 0.6 vs. 7.3 ± 2.0 × 104 cells/µm2, p < 0.05; Diaphragm: 4.2 ± 1.8 vs. 12.3 ± 3.2 × 104 cells/µm2, p < 0.001; TA: 1.5 ± 0.5 vs. 4.1 ± 0.7 × 104 cells/µm2, p < 0.0001) (Figure 3(a)–(f)). Similarly, UCP1-RFP positive cells were significantly increased in PRURD2 mice compared to PRURDBA controls (Heart: 0.05 ± 0.07 vs 1.82 ± 1.59 × 104 cells/µm2, p < 0.05; Diaphragm: 0.001 ± 0.002 vs 1.027 ± 0.302 × 104 cells/µm2, p < 0.0001; TA: 0.03 ± 0.07 vs 0.63 ± 0.40 × 104 cells/µm2, p < 0.01), and these cells colocalized with PDGFRα-GFP-positive cells. The percentage of UCP1-RFP positive cells among PDGFRα-GFP positive cells were also significantly increased in PRURD2 mice compared to PRURDBA controls (Heart: 0.62 ± 0.23 vs 0.84 ± 2.37%, p < 0.05; Diaphragm: 0.30 ± 0.54 vs 3.37 ± 1.29%, p < 0.0001; TA: 0.76 ± 0.35 vs 5.4 ± 2.09%, p < 0.0001). In the fibrotic regions, α-SMA-positive areas were observed, and PDGFRα-GFP positive cells were more densely distributed in PRURD2 mice (Figure 3(g)).
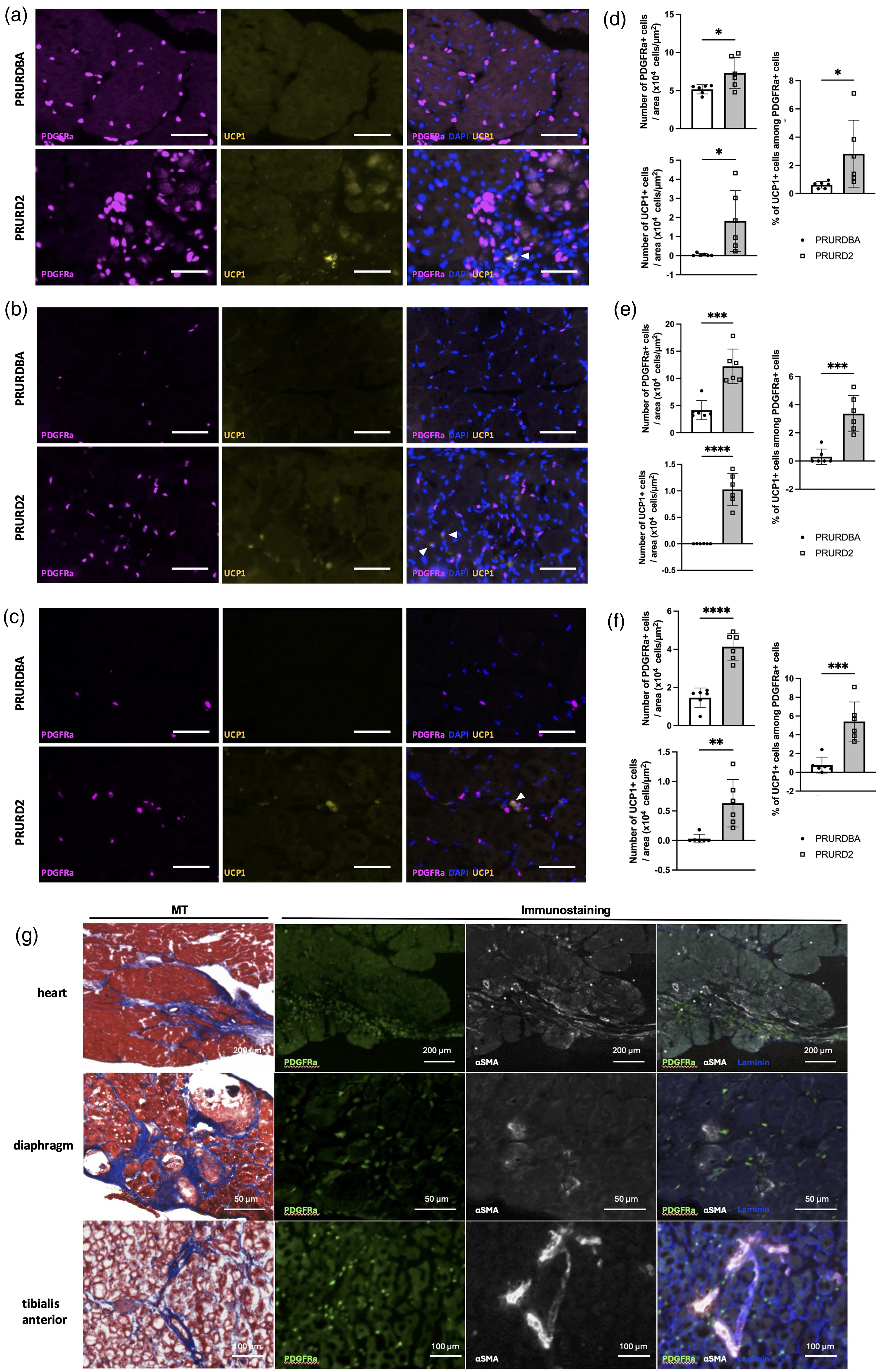
Immunohistochemical analysis of FAP abundance and BAT differentiation in cardiac and skeletal muscle of FAP-BAT double reporter D2-mdx (PRURD2) and DBA/2J (PRURDBA) mice. (a–f) Representative immunofluorescence images and quantification showing PDGFRα and UCP-1 expression in the heart (a, d), diaphragm (b, e), and tibialis anterior (TA) (c, f) muscles of PRURDBA and PRURD2 mice. White arrowheads indicate co-localization between PDGFRα and UCP-1 signals, consistent with FAP to BAT lineage transition (Scale bars:100 µm). (g) Representative Masson's Trichrome-stained and immunofluorescent-stained sections showing α-SMA (fibrotic marker) and laminin (basement membrane marker) in PRURD2 mouse heart, diaphragm, and TA muscles. Data are presented as mean ± SD with individual values. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, comparisons between groups as indicated.
Single-cell RNA-sequencing identifies distinct FAP subpopulations in cardiac and skeletal muscles from D2-mdx mice
Single-cell RNA sequencing of isolated FAPs from the heart, diaphragm, and TA muscles identified 11 transcriptionally distinct FAP subpopulations based on clustering of previous human FAP clusters, 23 characterized by enriched expression of known marker genes (Figure 4(a)–(c)). Cell population distribution analysis revealed an increased proportion of pre-fibrogenic FAPs, identified by the gene Cd55, in PRURD2 mice not only specific tissue but across all tissues compared to PRURDBA [Heart: 14.5% vs. 17.4%, Diaphragm: 18.2% vs. 29.5%, TA: 33.9% vs. 45.2%] (Figure 4(d)). Therefore, we used integrated data from the FAPs of all tissues to advance the analysis of the differentiation tendencies promoted by PRURD2. This Cd55 + population also expressed fibrogenic markers, including Wnt10b 24 and Adamts16. 25 (Figure 4(e) and (f)), supporting their role in promoting fibrosis.
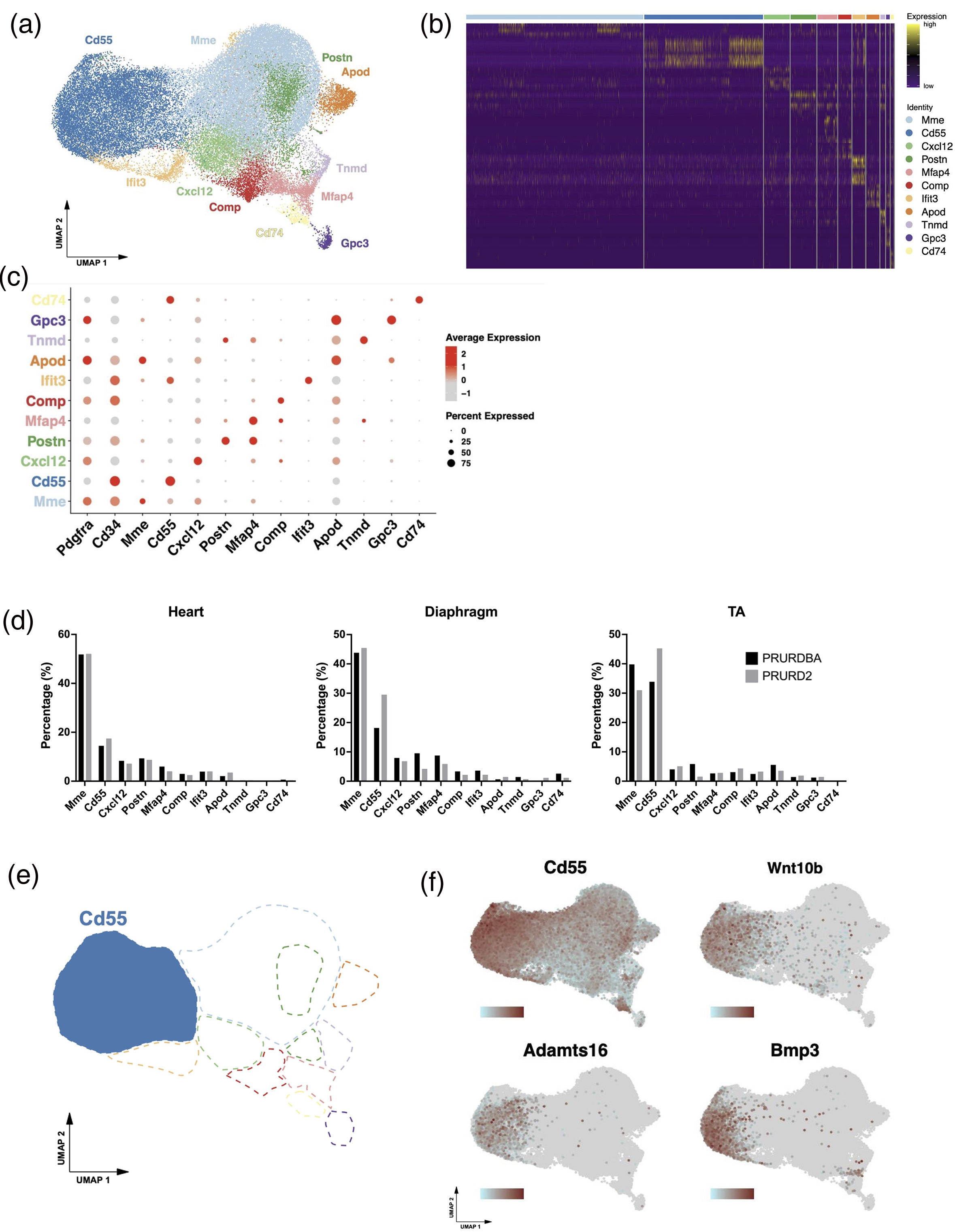
Single-cell RNA sequencing data of FAPs from FAP- BAT double reporter D2-mdx and DBA/2J mice. UMAP plot showing clustering of single-cell RNA-sequenced FAPs into 11 transcriptionally distinct subpopulations (a) with Heatmap (b), and dot plot illustrating expression levels and percent expression of selected markers (c) in PDGFRα-GFP tagged cells across all samples from FAP-BAT reporter D2-mdx and DBA/2J mice. (d) Cell population distribution plots comparing FAP subpopulation frequencies between PRURDBA and PRURD2 mice. UMAP plots showing the Cd55 + subpopulations associated with pro-fibrotic FAPs (e) and the expression plots showing the distribution and intensity of fibrosis-related gene expression (Cd55, Wnt10b, Adamts16, and Bmp3) within the Cd55 + cluster (f) in PDGFRα-GFP tagged cells across all samples from FAP-BAT reporter D2-mdx and DBA/2J mice. Darker red indicates higher expression levels.
To explore FAP differentiation trajectories, pseudotime analysis was initiated from Gpc3 + and Cd74 + progenitor clusters23,26 (Figure 5(a)), revealing unique lineages progressing toward Cd55+ (pre-fibrogenic) and Mme + (pre-adipogenic) states. Along the trajectory, we found increasing expression of lineage-specific surface markers for FAP subpopulations (Cd55 and Mme) and associated fibrogenic or adipogenic genes (Figure 5(b)), suggesting a structured and divergent differentiation program within FAPs in dystrophic muscle.
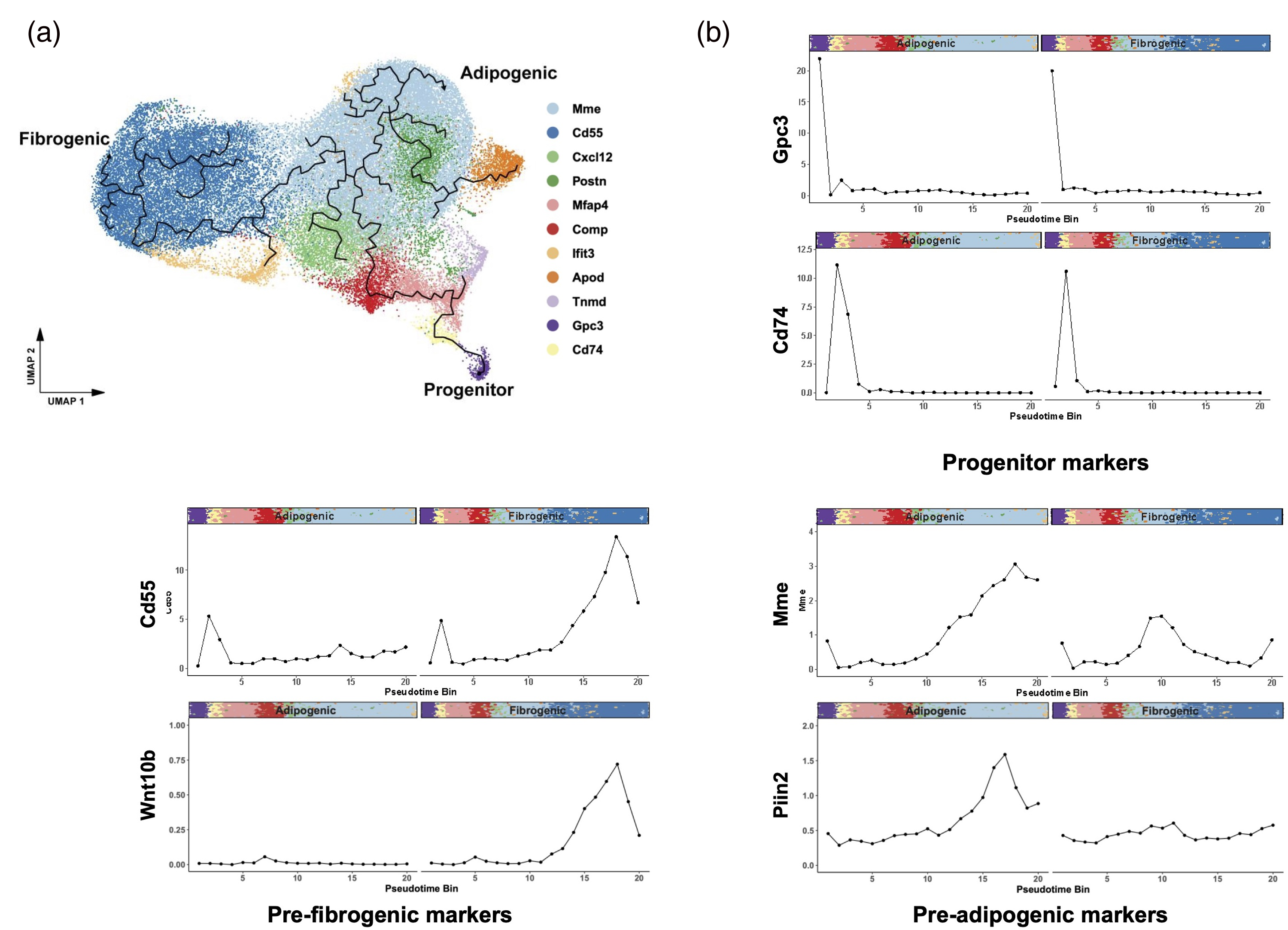
Pseudotime analysis of fibroadipogenic progenitors (FAPs) in all FAP-BAT double reporter D2-mdx (PRURD2) and DBA/2J (PRURDBA) mice. (a) UMAP plot of all sequenced FAPs overlaid with pseudotime trajectories, illustrating lineage progression from progenitor FAPs toward either fibrogenic or adipogenic in PDGFRα-GFP tagged cells across all samples from FAP-BAT reporter D2-mdx and DBA/2J mice. (b) Binned average expression of selected lineage-specific genes plotted along each pseudotime trajectory leading to fibrogenic or adipogenic fate. Colored bars indicate the cluster identity of each cell along the differentiation trajectory.
PRURD2 mice exhibit fibrosis-associated intercellular communication networks absent in PRURDBA
Analysis of intercellular communication patterns using CellChat identified distinctly altered signaling networks between PRURD2 and PRURDBA mice, particularly within FAP subpopulations. In PRURD2 mice, we observed enriched activity in key fibrotic signaling pathways, including collagen and TGF-β in the heart (Figure 6(a)), NOTCH and ANGPTL in the diaphragm (Figure 6(b)), and collagen and WNT in the TA muscle (Figure 6(c)). Notably, PRURD2 exhibited a marked increase in communication network among progenitor FAPs (Gpc3+/ Cd74+), suggesting enhanced cellular crosstalk driving fibrogenic progression. In addition, several signaling pathways were uniquely enriched in PRURD2 mice, with no detectable activity in PRURDBA controls, including EPHA, EPHB, NT, PCDH, CDH, SEMA5, PTPRM, SELE, GRN, and LIFR in the heart, EPHA, NT, VCAM, SEMA6, PTN, CSPG4, Glutamate, and ESAM in the diaphragm, and EPHA, EPHB, NT, VCAM, SEMA3, SEMA4, SEMA5, SEMA6, NT, CD39, Adenosine, AGT, Prostaglandin, THY1, PLAU, AGRN, and MMP in the TA muscle. These findings underscore the presence of fibrosis-associated and tissue-specific signaling interactions in PRURD2 mice that are absent in non-dystrophic controls, which could contribute to the exacerbated fibrotic phenotype. To further characterize molecular differences, differential gene expression analysis was performed using NOIseq. The top 20 upregulated genes in PRURD2 mice, ranked by log2 fold change, included genes involved in fibrogenic differentiation and extracellular matrix remodeling, including Cxcl13, Cxcl3, and Ly6d in the heart (Figure 6(d)); Klk1, Fgf23, Ly6d, Serpinb2, and Mmp13 in the diaphragm (Figure 6(e)); and Ccl17, Postn, and Adam12 in the TA muscle (Figure 6(f)). A full list of differentially expressed genes is provided in Supplemental data S1. We also combined the data from TA and Diaphragm FAPs as Skeletal muscle FAPs and compared them with Heart FAPs, referred to as Cardiac muscle FAPs. The top 200 genes with the largest changes, with -log10 (1-Probability) of 1 or more and log2 Fold Change of 1 or more, were extracted and compared between Skeletal muscle FAPs (TA and Diaphragm FAPs) and Cardiac muscle FAPs (Heart FAPs) in PRURDBA and PRURD2 mice (Supplemental data S2). Among the extracted genes, 30 distinct genes were identified between PRURDBA and PRURD2 mice. In PRURDBA mice, Skeletal muscle FAPs exhibited increased expression of Dclk3, which is involved in the innervation of skeletal muscles, and Kcna5, which is associated with the regulation of electrical excitability in muscle cells. In contrast, Cardiac muscle FAPs showed increased expression of stress response-related genes Chodl and Cryaa. On the other hand, in PRURD2 mice, Skeletal muscle FAPs demonstrated increased expression of inflammation-related genes Gpr174 and Vcam1, while Cardiac muscle FAPs exhibited increased expression of genes related to cardiac morphology and surface protection, such as Chip and Muc16.
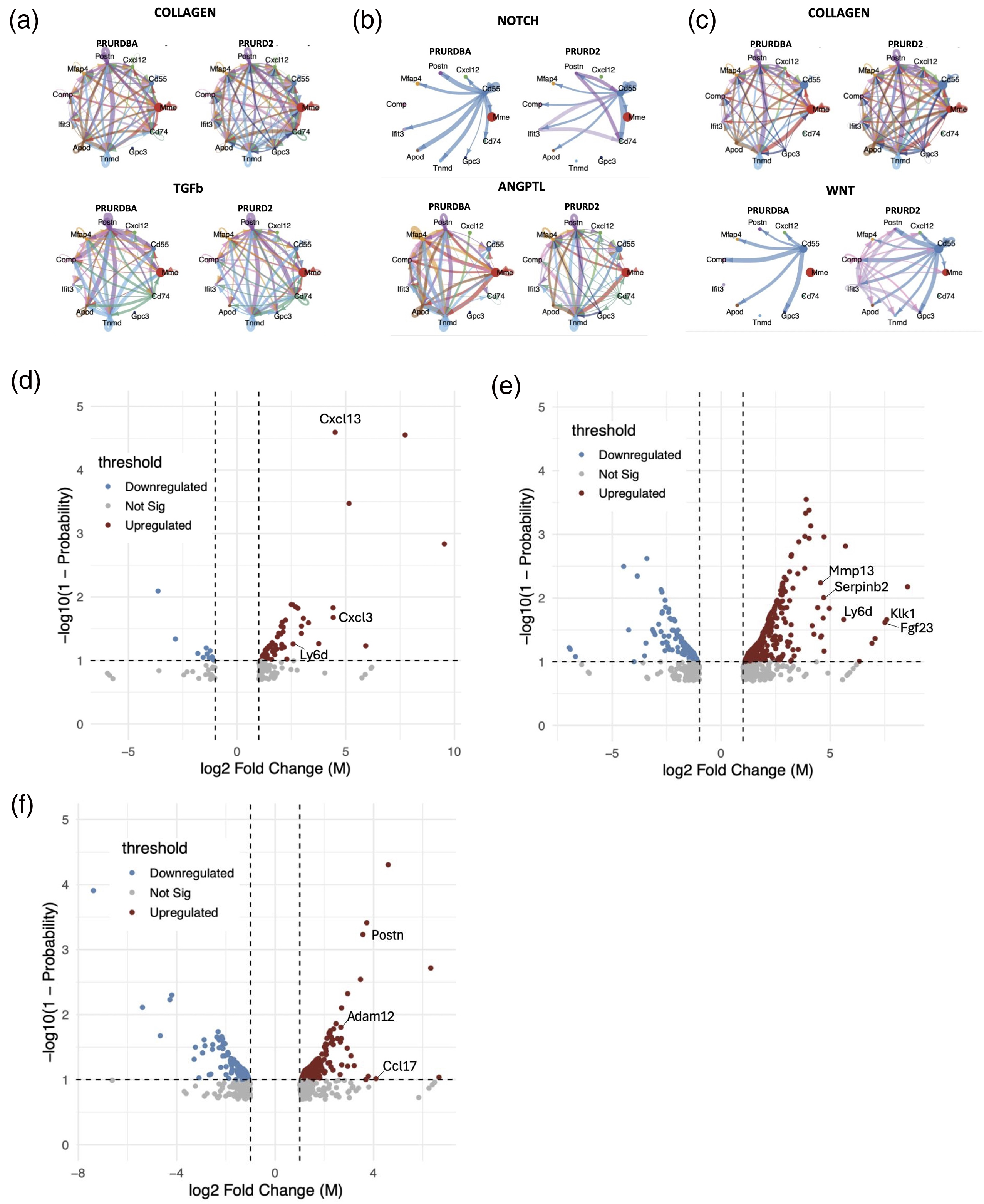
Cell-cell interactions and differential expression genes of fibroadipogenic progenitors (FAPs) across all subpopulations in FAP-beige adipose tissue (BAT) double reporter D2-mdx (PRURD2) and DBA/2J (PRURDBA) mice. Signal pathways exhibiting distinctly different cell communication patterns involving the progenitor FAP population (Gpc3 and CD74) between FAP-BAT double reporter DBA/2J (PRURDBA) mice and FAP-BAT double reporter D2 (PRURD2) mice in significantly upregulated pathways related to fibrogenesis among the heart (a), diaphragm (b), and tibialis anterior muscles (c). The names of genes involved in fibrogenic differentiation are plotted among the top 20 upregulated genes in PRURD2 compared to PRURDBA with -log10(1-Probability) of 1 or more and log2 Fold Change of 1 or more among the heart (d), diaphragm (e), and tibialis anterior muscles (f).
Discussion
In this study, we demonstrate that PRURD2 (FAP-BAT double reporter D2-mdx) mice develop severe fibrosis in both cardiac and skeletal muscle, characterized by an expansion of the FAP population and enhanced fibrotic differentiation. Single-cell RNA sequencing revealed transcriptional heterogeneity within FAPs, identifying intrinsically pro-fibrotic subpopulations, particularly the cd55 + cluster, as key drivers of fibrogenic degeneration in the D2-mdx model. These findings support a central role for FAP population expansion and their heightened fibrogenesis in the fibrotic pathology of DMD. While previous research has identified the role of FAPs in promoting fibrosis and adipogenesis in dystrophic muscle, most studies have relied on the BL10-mdx model, 27 which fails to fully recapitulate the extensive fibrosis observed in human DMD patients. Our work addresses this gap by using the D2-mdx background, which exhibits severe fibrosis comparable to human DMD, and introducing a novel FAP-BAT double reporter mouse model that enables in vivo tracking of FAP differentiation into fibrotic and beige adipose tissue lineages.
Appropriate animal models are essential for understanding disease mechanisms and developing effective therapies. While the C57BL/10ScSn-Dmdmdx/J (BL10-mdx) mouse is widely used, it exhibits only mild limb muscle pathology and minimal cardiac abnormalities, features that diverge from the human DMD phenotype. The D2-mdx strain, generated by backcrossing BL10-mdx onto a DBA/2J background, 19 displays more severe muscle degeneration and extensive fibrosis, making it a more suitable model for studying the fibrotic complications of DMD. 14 Utilizing this genetic background, we generated PRURD2 mice, a novel FAP-BAT dual-reporter model enabling in vivo tracking of FAP dynamics and BAT differentiation during the progression of muscle pathology in DMD. Van Putten's 2019 study on the natural disease course of D2-mdx mice reported that histological evaluations showed some improvements in pathological features at 34 weeks of age compared to 10 weeks of age, although individual variability was significant. 19 In our preliminary experiment investigating the survival curve of D2-mdx mice (n = 11), 20% of the mice died naturally over the course of 12 months (Supplementary Figure S4). Grip strength gradually declined and continued to decrease progressively up to 1 year of age. Based on these findings, we decided to sample 1-year-old D2-mdx mice in this study, as sufficient pathological changes are expected to occur in the heart, diaphragm, and skeletal muscles by this time point. It has been reported that the DBA2 mouse strain carries a mutation in the Ltbp4 gene, which may affect muscle function and regeneration.28,29 Therefore, we have confirmed that PRURDBA and PRURD2 have properly inherited the Ltbp4 gene (Supplementary Figure S5).
To confirm the DMD-like phenotype in PRURD2 mice, we assessed muscle strength and structure. Despite no significant differences in body weight, grip strength was markedly reduced in PRURD2 mice. These results contrast prior studies at earlier time points that showed no significant difference in normalized grip strength. 19 Our findings at 12 months of age suggest that aging accelerates muscle decline, highlighting the utility of long-term studies in capturing disease progression. Interestingly, this reduction in muscle strength occurred in the absence of changes in body or muscle weight between PRURD2 and PRURDBA mice, consistent with previous observations of age-related muscle deterioration even in the DBA/2J background. 30 Herein, DBA mice showed a significant decline in body weight, muscle mass, and strength by 52 weeks of age compared to 24 weeks. 30 Similarly, in a study examining the natural disease progression of D2-mdx mice, male DBA controls weighed over 30 grams at 32 weeks, 19 whereas in our study, 1-year-old PRURDBA mice averaged 25 grams. These findings indicate a progressive deterioration in muscle quality in DBA mice between 6 and 12 months of age, which likely explains the lack of significant difference in muscle mass between PRURDBA and PRURD2 mice despite profound functional impairment in the latter.
Histological analysis revealed that fibrosis was diffuse and uniform in the diaphragm and TA of PRURD2 mice, whereas in the heart, it was confined to the right ventricular pericardium, a distribution previously observed in D2-mdx hearts. 31 Clinically, right ventricular failure in DMD is often attributed to pulmonary complications, but increasing evidence links it to direct cardiac dysfunction in DMD patients, including arrhythmia, shock, and acute heart failure. 32 The prominent ventricular fibrosis observed in PRURD2 mice suggests this model may also serve to investigate the pathogenesis of right heart failure in DMD. The gastrocnemius muscle has been reported to exhibit more severe fibrosis. 19 However, in this study, we selected the tibialis anterior muscle, prioritizing its high reproducibility and the extensive experience with its evaluation at our facility.
Using the PDGFRa and UCP1 dual-reporters, we identified increased FAPs and BAT signals in the heart, diaphragm, and TA muscles of PRURD2 mice, with co-localization in fibrotic regions. These findings align with previous reports in both murine and human DMD muscle.10,33,34 Our mouse model uniquely allows in vivo tracing of FAPs undergoing spontaneous BAT differentiation, a process we previously observed during muscle regenerative responses following injury. 35 It is already known that fat infiltration (white adipose tissue) increases in DMD mice; however, our results support that FAP-BAT differentiation is also enhanced in PRURD2 mice. This BAT differentiation may reflect an intrinsic repair mechanism that is overwhelmed in the chronic muscle damage environment of DMD.
In our previous study with human FAPs, we identified a Cd55 + fibrogenic subpopulation induced by chronic TGF stimulation. 23 Here, we confirmed that Cd55 as a reliable and conserved marker of pro-fibrotic FAPs in the D2-mdx mouse model. This population expressed fibrosis-related genes, including Wnt10b and Adamts16, further supporting its functional role. Previous studies have identified FAP clusters marked by Cthrc1 and Vcam1 in the quadratus muscle of mdx5cv mice, 10 and others have reported upregulated fibrotic genes in the DMD patient muscle. 36 Our findings reinforce the potential of Cd55 as a universal marker of fibrogenic FAPs across species. Ongoing work in our lab is testing this hypothesis in human DMD muscle biopsies.
We have previously reported that FAP-mediated cell-cell signaling is specific to the underlying degenerative pathology. 23 In this study, CellChat analysis revealed a pronounced tissue-specific expansion of fibrotic signaling networks in PRURD2 mice, particularly involving progenitor FAPs (Cpc3+/Cd74+). Among the pathways significantly upregulated in PRUD2 tissues were several canonical fibrogenesis signaling cascades, including collagen, 37 TGF-β, 38 NOTCH, 39 ANGPTL, 40 and WNT. 41 Importantly, we also identified signaling pathways uniquely upregulated in PRURD2 mice and absent in PRURDBA controls, including THY1, 42 TWEAK, 43 EPHA and EPHB, 44 and SEMA6, 45 suggesting a disease-specific reprogramming of the FAP signaling microenvironment in response to chronic muscle degradation. Among these, EPHA2 emerged as a common mediator, interacting directly with the TGF-β fibrotic axis, 46 functioning as a receptor for WNT ligands, 47 and regulating Notch signaling. 48 Transcriptomic analysis further revealed upregulation of several TGF-β-responsive genes associated with fibroblast activation and tissue remodeling in PRURD2 mice, including Cxcl13 (a chemokine induced by TGF-β and known to drive fibroblast differentiation and fibrosis, 49 Mmp13 (a matrix metalloproteinase upregulated during pathological tissue remodeling and modulated by TGF-β/BMP signaling, contributing to fibroblast differentiation and ECM remodeling 50 ), Ccl17 (a chemokine known to interact with its receptor CCR4 on fibroblasts, activating TGF-β/Smad signaling to promote fibroblast activation and tissue fibrosis, 51 and Postn (a matricellular protein that plays a critical role in the expression of collagen I and α-SMA induced in response to TGF-β1 stimulation in primary fibroblasts. 52 Collectively, these results highlight a complex interconnected pro-fibrotic signaling network in PRURD2 mice, driven by both canonical and some novel pathways. Targeting these pathways and their fibrogenic effector genes offers promising therapeutic strategies to reduce muscle fibrosis in DMD.
This study has several limitations. First, our analysis was restricted to a single time point (12 months of age), which limits our ability to capture the temporal dynamics of FAP differentiation and fibrogenesis throughout disease progression. Future studies incorporating longitudinal sampling will be essential to delineate the trajectory of fibrotic remodeling and FAP lineage plasticity in DMD. Second, while the UCP-1 reporter allowed the visualization of BAT differentiation, no in-depth analysis of BAT or white adipose tissue was performed. Furthermore, our study focused on transcriptional profiling and did not include protein-level validation of key differentially expressed genes or pathway activity. Despite these limitations, the primary aim of this study was to establish and characterize the transcriptional landscape of FAPs across multiple dystrophic tissues using a genetically tractable reporter model. Future studies will incorporate interventional experiments and targeted validation studies to assess the functional consequences of specific FAP subpopulations and signaling pathways in the progression of DMD-related muscle fibrosis.
In conclusion, we developed and characterized a novel FAT-BAT double reporter D2-mdx mouse model (PRURD2) that recapitulates the severe skeletal and cardiac muscle fibrosis observed in DMD. In this model, FAPs exhibit activation of pro-fibrotic signaling pathways, mirroring key pathological features of the human disease. The PRURD2 mice offer a valuable model to further study the cellular and molecular mechanisms of fibrosis and holds promise for developing new anti-fibrotic therapies for DMD.
Supplemental Material
sj-xlsx-1-jnd-10.1177_22143602261441956 - Supplemental material for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse
Supplemental material, sj-xlsx-1-jnd-10.1177_22143602261441956 for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse by Hiroyori Fusagawa, Justin Lau, Sankalp Sharma, Mengyao Liu, Yusef Samimi, Gabriel Franchet-Schaer, Ashley Fang, Minami Fusagawa, Hubert Kim, Brian T Feeley and Xuhui Liu in Journal of Neuromuscular Diseases
Supplemental Material
sj-xlsx-2-jnd-10.1177_22143602261441956 - Supplemental material for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse
Supplemental material, sj-xlsx-2-jnd-10.1177_22143602261441956 for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse by Hiroyori Fusagawa, Justin Lau, Sankalp Sharma, Mengyao Liu, Yusef Samimi, Gabriel Franchet-Schaer, Ashley Fang, Minami Fusagawa, Hubert Kim, Brian T Feeley and Xuhui Liu in Journal of Neuromuscular Diseases
Supplemental Material
sj-docx-3-jnd-10.1177_22143602261441956 - Supplemental material for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse
Supplemental material, sj-docx-3-jnd-10.1177_22143602261441956 for Fibrotic differentiation profile of skeletal and cardiac muscle fibroadipogenic progenitors in D2-mdx mouse by Hiroyori Fusagawa, Justin Lau, Sankalp Sharma, Mengyao Liu, Yusef Samimi, Gabriel Franchet-Schaer, Ashley Fang, Minami Fusagawa, Hubert Kim, Brian T Feeley and Xuhui Liu in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
The authors wish to thank for Dr Steven Garcia (UCSF) for his guidance of scRNAseq.
The UCSF Genomics CoLab for assistance with scRNAseq experiments.
ORCID iDs
Ethics approval and consent to participate
All animal procedures were performed in compliance with the institutional Animal Studies Subcommittee (IACUC) (ACROP # 24-003).
Consent for publication
Not applicable.
Author contributions
Conceptualization: X.L.; Content Refinement: H.F. and X.L.; Manuscript Writing: H.F.; Manuscript
Editing: all authors, but primarily H.F., J.L. and X.L.; Experimental work: H.F., Y.S., A.F., and M.F.; Data analysis: H.F., Y.S., and J.L.
All authors approved the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by grants from Department of Defense (DMDRP grant #MD230014) and Muscular Dystrophy Association (MDA, research grant #MDA 957140).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Single-cell RNA sequencing data are available in Gene Expression Omnibus (GEO) under accession number “GSE322960”. All other data that was collected and used to draw conclusions from this research is available and can be obtained by contacting the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.