
Abstract
Introduction:
Spinal muscular atrophy (SMA) is a neuromuscular disease that affects patients and caregivers worldwide, including in India, with a significant economic burden.
Methods:
A comprehensive literature review was conducted to evaluate SMA, patient journeys, healthcare access, and treatment barriers in India, forming the basis for an expert panel discussion.
Results:
The experts highlighted that early detection of SMA through newborn, carrier, or prenatal screening, and diagnosis through genetic testing enable timely interventions including disease-modifying therapies (DMTs) and multidisciplinary care. Currently, in India, Risdiplam is the only Drugs Controller General of India (DCGI) approved DMT for SMA. It is administered orally and approved for use in SMA type 1, 2, 3, and 4 among pediatric and adult patients. The management of SMA often revolves around the management of its complications, which requires respiratory, nutritional, and orthopedic care, both while awaiting and after receiving DMTs. Therefore, the gold standard for SMA care requires the availability of multidisciplinary care involving specialists from various fields. Despite the availability of DMTs, challenges such as affordability and timely access to these therapies remain major hurdles faced by SMA patients in India. Additionally, a lack of awareness among healthcare professionals, contributed to underdiagnosis and undiagnosed cases, further exacerbating the situation.
Conclusion:
This review provides insights from the expert opinions of Indian pediatricians, neurologists, geneticists, pediatric neurologists, and pediatric pulmonologists on the burden of SMA, diagnosis and management practices, importance of a multidisciplinary approach, challenges faced in SMA care in India, and strategies to overcome these challenges.
Introduction
Rare or orphan diseases are conditions that affect a very small number of people (≤10 per 10,000 population) and are often associated with inappropriate management, adverse health outcomes, chronic debilitation, and early death.1,2 The global point prevalence of rare diseases is estimated to be 3.5%–5.9%. 3 According to the Indian Organization of Rare Diseases, approximately 450 rare diseases have been identified in India, with a significant number being neuromuscular disorders.4,5
Spinal muscular atrophy (SMA), a neuromuscular disease, is a rare disease inherited in an autosomal recessive mode. SMA is classified into types 0, 1, 2, 3, and 4 depending on the time of its onset and severity; in type 0 SMA, the manifestation begins in utero, and the affected infants usually die within the first few weeks of life, while in type 4 SMA, patients can lead almost normal lives. The disease is characterized by the degeneration of anterior horn cells in the spinal cord, leading to the destruction of alpha motor neurons, which clinically manifest as proximal muscle weakness and atrophy. 6 According to the updated 2024 National Organization for Rare Diseases (NORD) data, the global incidence of SMA is estimated to be 1 in 10,000 live births affecting both males and females equally. 7 Although SMA is considered relatively common among rare diseases in India, there is a significant lack of data on its incidence and prevalence among the Indian population. 8
The disease is deeply concerning due to its significant impact on the quality of life (QoL) of patients and their caregivers. Moreover, it imposes a substantial economic burden and leads to progressive motor difficulties, including sitting, standing and walking. These impair daily activities and increase dependance on caregivers. 9 According to a systematic review, SMA puts a significant burden on caregivers, leading to impaired QoL, financial stress, diminished ability to work, and low productivity, as it requires the caregivers to devote a significant amount of time to informal care. 10 Without effective management and appropriate therapies, the likelihood of type 1 SMA patients surviving to 1 year of age is reported to be 40%, while the probability of reaching 20 years of age drops to just 0%. 11 Although the survival probability of children with type 2 SMA (52%–100%) is good, there is a lack of quality of life, as many eventually lose their ability to sit and develop feeding difficulties along with severe respiratory problems.11,12 These statistics indicate that early treatment is necessary to prevent disease progression and improve survival outcomes.
The United States Food and Drug Administration (US FDA) has approved three disease-modifying therapies (DMTs), namely, Nusinersen (in 2016), Onasemnogene abeparvovec (in 2019), and Risdiplam (in 2020 for patients ≥2 months of age, in 2022 for patients <2 months of age) for managing SMA. Of these, only Risdiplam is approved by the Drug Controller General of India (DCGI) in India among pediatric and adult patients with SMA. 13 Nusinersen is an antisense oligonucleotide therapy administered via intrathecal injection across all ages and Onasemnogene abeparvovec is a gene therapy administered via intravenous infusion up to 2 years age/ less than 21 kg. Risdiplam is the only DMT administered orally. 5 The effectiveness and safety of these therapies are well documented in the ENDEAR and CHERISH trials (for Nusinersen);14,15 START and STRIVE trials (for Onasemnogene abeparvovec);16,17 and FIREFISH, SUNFISH, RAINBOWFISH, and JEWELFISH trials (for Risdiplam).18–22 Despite these therapies being available to manage SMA, their accessibility and affordability are the biggest challenges for SMA patients in India. Nusinersen and Onasemnogene abeparvovec are available only through the Individual Patient Humanitarian Access Program/ global access program for rare diseases, whereas Risdiplam is commercially available in the country. Additionally, the Government of India, under the National Policy of Rare Diseases (NPRD) 2021 provides financial support of 50 lakhs for patients with any rare disease, including SMA. 23 However, the funding provided is not sustainable, considering the long-term management of SMA. 5
Considering this scenario, it is crucial to reassess and reevaluate the role of all stakeholders including patients/parents, parent support groups, multidisciplinary health care professionals, pharma, government, employee's health schemes, research funding institutions, crowd funding platforms etc. in management and explore innovative approaches to enhance the QoL and improve the survival likelihood for SMA patients (IAMG 2023). 24 This study was undertaken to understand the opinions of experts in managing SMA regarding its burden, diagnosis, management strategies, challenges encountered, and the way forward in the Indian context.
Methods
An extensive search was conducted in electronic databases to gather existing evidence on SMA, the patient journeys of those with SMA, access to healthcare services, and treatment barriers in the management of SMA in India (Table S1). Based on this literature review, a discussion guide was developed for an invited panel. A panel of experts was invited to provide insights on the management of SMA in India. Panel members were selected through purposive sampling, based on their clinical experience in treating SMA, academic involvement, and affiliations with leading hospitals or medical institutions. The panel included pediatricians, neurologists, geneticists, pediatric neurologists, and pediatric pulmonologists. While the panel did not include all SMA-treating physicians in the country, it was designed to ensure diverse perspectives from key specialties involved in SMA care.
A total of six virtual meetings and panel discussion were conducted, focusing on the characteristics of SMA, the current treatment landscape of SMA, and the role of Risdiplam in managing SMA in India. The discussion guide for the meeting is presented in (Table 1). The following sections summarize expert opinions based on the views and statements expressed by the panelists during the meetings and discussions.
A discussion guide for the advisory board meeting.
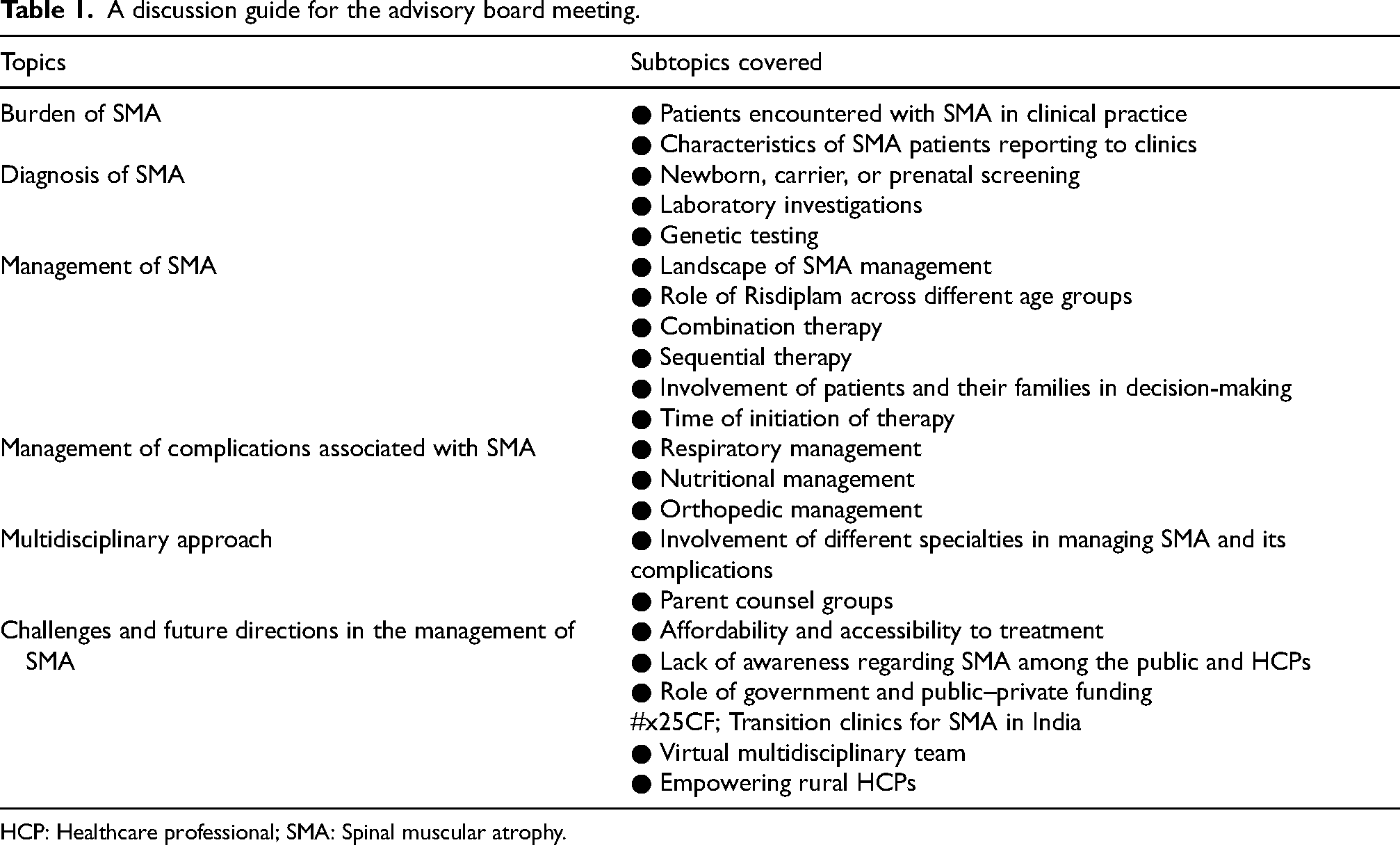
HCP: Healthcare professional; SMA: Spinal muscular atrophy.
Characteristics and burden of SMA
SMA, an inherited neuromuscular disease, results in the loss of nerve cells in the anterior horn cells due to a deficiency of the survival motor neuron (SMN) protein. This deficiency is caused by the absence of the SMN1 gene, typically due to homozygous deletions or mutations in the exon 7 of the SMN1 gene located on chromosome 5q13. While both the SMN1 and SMN2 genes produce the same SMN protein, SMN1 generates 90% of the functional protein required for neuromuscular function, whereas SMN2 produces only 10%. 25 In type 0 SMA, symptoms begins in utero. In type 1 SMA patients (non-sitters), symptoms manifest within the first 6 months of life. These include limited head control, hypotonia, areflexia, bell-shaped chest, and tongue fasciculations. In type 2 SMA patients (sitters), symptoms typically manifest between 6 and 18 months of age and include hypotonia, areflexia, scoliosis, and progressive proximal weakness that initially affects the legs more than the arms. In type 3 SMA patients (walkers), symptoms manifest around 18 months of age with progressive proximal weakness. Type 4 SMA presents in adulthood with mild leg weakness that progresses to proximal weakness. 6
In the Indian population, the carrier frequency of SMA among healthy individuals with no family history was reported to be 1 in 38 (2.6%), while among individuals with a positive family history, it was 1 in 31 (3.2%). 8 As per data from patient advocacy groups Cure SMA and ORDI (Organization for Rare Diseases in India), the current confirmed diagnosed cases in India are expected to be 1230, and the data from the various Centre of Excellence for Rare Disease in India estimate a similar number. 26 While published data on mortality rates for SMA subtypes in India are currently lacking, global data report type 1 SMA to be the most prevalent type with high mortality rates (67.7%), followed by type 2 SMA (with a mortality rate of 11%) 12 (Chan SHS et al. 2023). In the absence of ventilator support, respiratory failure is the primary cause of mortality in type 1 and type 2 SMA patients. 6
Parents of patients with SMA and older children with SMA experience high levels of anxiety and depression.27–29 A cross-sectional study reported anxiety among 40% and depression among 25.2% school-going SMA children aged 8–18 years. 27 A cross-sectional study from North India reported significantly elevated infant behavior questionnaire-revised (IBQ-R) scores among SMA patients aged 3 months–5 years for sadness, distress to limitation, rate of recovery from distress, and fear as compared to healthy controls; these patients also scored lower for soothability compared to healthy controls. 28 Denial is another significant characteristic that has been observed among parents of children with neuromuscular diseases. A study reported that parents often deny their child's condition for an unusually long period and, upon acceptance, experience feelings of helplessness and chronic guilt. 29 The opinions of the experts in the present study align with those of the reported literature and indicate that SMA is a burden affecting not only the patients but also their families (Table 2).
Expert opinion on the burden and characteristics of SMA.

AIIMS: All India Institute of Medical Sciences; SMA: Spinal muscular atrophy.
Screening and diagnosis of SMA
Newborn screening
Early detection through newborn, carrier, or prenatal screening enables timely interventions, preventing complications, significantly reducing long-term healthcare expenses, and improving patient outcomes.30–32 These measures align with national policies that promote preventive healthcare and early intervention and also aid in increasing awareness among the public and healthcare professionals (HCPs) regarding the significance of early screening and prompt treatment. 31 However, routine newborn screening is not part of any national screening policy or is commonly practiced in India due to a lack of well-established, easily accessible curative treatments for the patients. 5
Population carrier screening
The objectives of these screening tests are to identify carriers and provide them with reproductive options, such as choosing to marry someone who does not carry the same disease and preventing the birth of an affected child.32,33 With increasing cases of rare diseases among couples without a familial history of rare diseases, an ideal approach would involve screening all couples for genetic disorders. 30
Early screening and diagnosis: Gold standard and real-world situation in India in comparison to high-resource settings
Experts mentioned that in clinical practice, type 1 patients are usually brought to the clinics at the age of 4–6 months due to symptoms such as difficulty in neck holding and feeding, and the presence of recurrent respiratory issues. However, due to poor early diagnosis and high early mortality, many type 1 SMA patients may not reach specialized clinics. As a result, the usual age of presentation particularly for patients with type 2 SMA remains beyond 2 years of age at clinics.
Screening and diagnosis of SMA are crucial because the cost of prevention through screening for single-gene disorders is much lower than the cost of treatment. 30
In India, molecular testing for SMA is the third most common test performed (after molecular testing for β-thalassemia and Duchenne muscular dystrophy); around 1052 prenatal and 2256 diagnostic molecular tests were performed to detect SMA from 1997 to 2020. 34
For diagnosis, a genetic test using blood samples can detect mutations or deletions in the SMN1 gene, which are indicative of SMA types 1, 2, and 3 in 95% of cases, and can also identify carriers. 35 The gold standard for confirming SMA through genetic testing is a quantitative analysis of SMN1 and SMN2 genes using quantitative polymerase chain reaction, multiplex ligation-dependent probe amplification, or next-generation sequencing. 36 If the SMN1 gene is not implicated or if clinical symptoms do not align with SMA, further diagnostic measures may include electromyography to assess muscle function, nerve conduction velocity studies to evaluate nerve signal transmission, and muscle biopsy for diagnosing other neuromuscular disorders. 35 The lack of awareness among HCPs regarding screening and diagnosis for SMA leads to misdiagnosis and delayed diagnoses, which affect the journey of SMA patients. 37 The opinions provided by the experts on screening for and diagnosis of SMA align with the reported literature (Table 3).
Expert opinion on the screening and diagnosis of SMA in India.

CPK: Creatine phosphokinase; SMA: Spinal muscular atrophy.
Management of SMA
Pharmacological intervention
Onasemnogene abeparvovec offers the potential advantage of a single-dose treatment compared to the other SMA treatments, which require multiple doses. However, acute liver toxicity, the need for prolonged use of steroids, and a lack of long-term data on its safety and efficacy in older and heavier patients often require patients to continue with other DMTs. 38 Furthermore, Nusinersen needs to be delivered intrathecally, which is a challenge in patients with scoliosis. It also requires hospitalization and can lead to infections. Risdiplam is the only DMT that can be taken orally with ease of administration at home and has proven long-term data across all SMA types. Despite their differing molecular mechanisms, optimal levels of SMN induction can be achieved with any of the three currently available DMTs, particularly when administered early. 38
However, Risdiplam holds significant potential for SMA management in countries such as India. In addition, four major clinical trials on Risdiplam have documented its effectiveness and safety profile in great detail among SMA patients of varying ages (Figure 1).18–22 Furthermore, a systematic literature review funded by F. Hoffmann-La Roche Ltd and conducted by authors affiliated with Roche or its contracted partners reported significant improvements in survival and motor function, as well as reduced likelihood of serious adverse events with Risdiplam compared to Nusinersen in type 1 SMA patients, based on a matching-adjusted indirect comparison. 39 While informative, such comparisons have methodological limitations and should be interpreted with caution. The expert opinions on the management of SMA in India are provided in (Table 4).

Clinical trials on the safety and effectiveness of Risdiplam.18–22 AE: Adverse events; CHOP-INTEND: Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; COVID-19: Coronavirus disease 2019; PK/PD: Pharmacokinetics/pharmacodynamics; SAE: Serious adverse events; SMA: Spinal muscular atrophy; SMAIS-ULM: Spinal muscular atrophy independence scale-upper limb module; WHO: World Health Organization.
Expert opinions on the management of SMA in India.

CGHS: Central Government Health Scheme; DMT: Disease-modifying therapy; ESI: Employees’ State Insurance Scheme; NGO: Non-government organization; SMA: Spinal muscular atrophy.
The dosage of Risdiplam, Nusinersen, and Onasemnogene abeparvovec along with their side effects are detailed in (Table 5)40–43

CNS: Central nervous system; SMN: Survival of motor neuron.
Holistic multidisciplinary care
Multidisciplinary care is the gold standard and state-of-the-art model for managing many severely debilitating neuromuscular diseases. 44 This model is especially effective because patients with these conditions often face a range of challenges, including significant muscle weakness and various extramuscular symptoms. By involving multiple specialties in a coordinated approach, multidisciplinary care ensures comprehensive management of these complex clinical presentations and allows patients to address numerous aspects of their condition in a single visit. 44 Considering the manifestations of SMA and its complications, a multidisciplinary team should consist of specialists such as a neurologist, pediatrician, pulmonologist, cardiologist, endocrinologist, orthopedic surgeon, gastroenterologists, speech therapist, nutritionist, geneticist, psychiatrist, neuropsychologist, and palliative care and hospice.36,44 It is also recommended that the coordination between the departments is primarily done by the neurologist or pediatric neurologist. 36
According to a report by the Ministry of Health and Family Welfare (MoHFW), Government of India, 12 centers of excellence staffed with experts from multiple disciplines have been identified, where SMA patients can avail treatment. 45 Furthermore, five Nidan Kendras have been set up for patients requiring genetic counseling and testing.23,26 Patient advocacy groups such as ORDI and CureSMA organize multidisciplinary clinics at regular intervals in various centers in India to facilitate SMA care and prevent complications, such as contractures. 46
Along with a multidisciplinary approach, parent counsel groups can be extremely beneficial for families of SMA patients. Advice from parents of a child with SMA can provide valuable insights into the important aspects of caring for a child with SMA. Additionally, the tips shared by these parents can be beneficial for HCPs, civil society organizations, and friends and families supporting those affected by SMA. 47 The opinions provided by the experts in the present study highlight the difficulties of patients with SMA in visiting multiple clinics and the importance of multidisciplinary teams and parent-to-parent counseling in managing SMA (Table 6).
Expert opinions on a multidisciplinary approach to managing SMA.
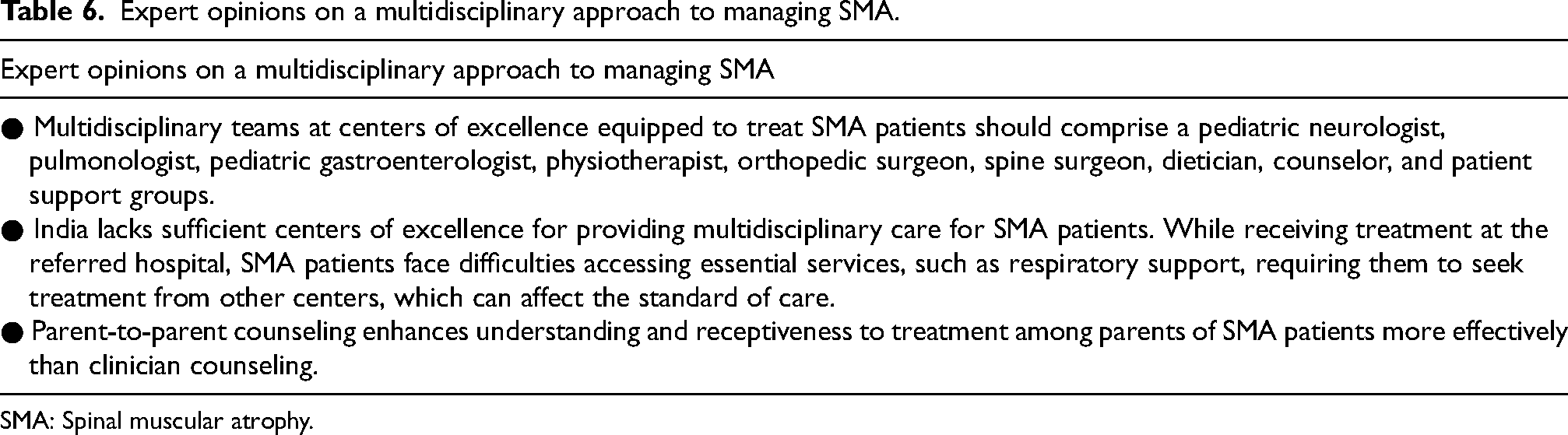
SMA: Spinal muscular atrophy.
Sequence of therapy and role of combination therapy
According to the Cure 2023 SMA community update survey report, combination therapy involves receiving more than one DMT simultaneously; this includes further treatment with either Nusinersen or Risdiplam after gene replacement therapy. Sequential therapy involves receiving more than one DMT at different times, such as administering gene replacement therapy after stopping other DMTs. 48
Reasons for sequential and combination therapy
A survey by Belter L et al. (2024) 48 reported various reasons for opting for sequential and combination treatments. For sequential treatment, the reasons included loss of function (which occurs in 12% of pediatric SMA patients and 27.3% of adult SMA patients), wanting a different DMT (children: 72.6% and adults: 88.0%), physician recommendation (children: 40.8% and adults: 17.8%), and wanting all possible DMTs (children: 39.6% and adults: 33.8%). 48
Challenges in categorizing combination therapy
Currently, there is no structured method to categorize the various treatment combinations for SMA patients using DMTs, making it challenging to analyze and understand the benefits and risks of different treatment plans, including regimens with single gene replacement therapy. 49
Evidence on analyzing treatment combinations
Researchers have attempted a few combinations to improve the QoL of SMA patients. A case series on 4 patients with type 1 SMA initially treated with Onasemnogene abeparvovec reported that the patients experienced a perceived plateau in therapeutic benefit. However, after initiation of Risdiplam, all patients demonstrated improvements in subjective and objective symptoms. 50
Comparison of combination and monotherapy
Mirea A et al. 51 reported that patients receiving the combination of Nusinersen and Onasemnogene abeparvovec had comparable motor function trajectories to those on monotherapy. The study also showed that motor function improvement was notably better in the 6 months following the first therapy compared to the 6 months after adding the second treatment. In addition, the study indicated that early initiation of therapy may be more crucial for improvement in symptoms than using a combination therapy. 51
The experts in the present study also expressed varying opinions on the use of combination/sequential therapy mirroring the diversity found in the existing literature (Table 7).
Expert opinion on the use of combination therapy in the management of SMA in India.

DMT: Disease-modifying therapy: SMA: Spinal muscular atrophy.
Prevention and management of complication in SMA
Prevention of complications of SMA can be done through early diagnosis, initiating treatment as early as possible within the first 3 months of life—when maximum motor neurons are dying, and by ensuring regular monitoring. Complications that SMA patients may develop include restrictive lung disease, sleep difficulties, scoliosis, poor weight gain with growth failure, and joint contractures. In addition, nutritional complications, such as bulbar dysfunction, are prevalent in type 1 SMA patients, however, they develop progressively in type 2 SMA patients and usually emerge late in life in type 3 SMA patients. 52 Gastrointestinal issues, such as delayed gastric emptying, constipation, and severe life-threatening gastroesophageal reflux, are also very common in SMA patients, which may lead to growth failure. However, a gastrostomy tube can be placed to manage these issues in such patients. 52 Conversely, non-ambulatory patients with SMA type 2 or 3 are at risk of developing obesity, which, in turn, increases the likelihood of associated conditions such as high blood pressure, metabolic syndrome, and diabetes. 20
Children diagnosed with SMA type 1 and 2 experience progressive deterioration in pulmonary function, characterized by the weakening of respiratory muscles, reduced lung and chest wall compliance, and diminished alveolar multiplication. 52 Respiratory failure is the leading cause of death in SMA type 1 and 2 patients, stemming from impaired cough, insufficient clearance of airway secretions, sleep hypoventilation, and recurrent pneumonia. Airway clearance techniques and noninvasive ventilation such as bilevel positive airway pressure (BPAP) are commonly used to manage respiratory insufficiency in these patients. 52 Additionally, due to the increased risk of pneumonia in SMA patients with respiratory muscle weakness, immunization with pneumococcal vaccinations should be prioritized. 53
The orthopedic complications experienced by SMA patients include joint contractures, scoliosis, and hip dislocation or subluxation. Scoliosis develops in nearly all children diagnosed with type 1 and 2 SMA. 54 Around 50% of these children develop spinal curvatures greater than 50°. Additionally, non-ambulatory individuals may experience thoracic kyphosis, which can affect lung function, while severe scoliosis may reduce cardiac output.20,52 According to the European Neuromuscular Centre Standard of Care Orthopaedic Working Group, surgical intervention is recommended for managing hip dislocation and contractures in patients with impaired function. For spine deformities, guided growth instrumentation is beneficial for skeletally immature patients, and posterior spinal fusion is recommended in skeletally mature patients; in addition, rib-based constructs may be employed to address parasol rib deformities. 55 As a result, a significant portion of SMA management focuses on addressing its complications, both before and after DMT becomes accessible to patients, to enhance the effectiveness of the treatments. The opinions of the experts strongly support the prevention and management of complications in SMA patients (Table 8).
Expert opinions on the management of complications associated with SMA.

BPAP: Bilevel-positive airway pressure; DMT: Disease-modifying therapy; PEG: Percutaneous endoscopic gastrostomy; SMA: Spinal muscular atrophy.
Challenges and future directions in the management of SMA
Global challenge
Though DMTs aid in managing SMA, the cost of the treatment poses the most significant challenge for patients and their families. Managing SMA complications further increases overall costs. Real-world data shows that the mean cumulative overall costs per SMA patient (from birth to 18 years of age) were substantial; 935,570 USD (6,92,35,610 INR) in the overall SMA group, 2,393,250 USD (17,72,95,625 INR) in type 1 SMA patients, 413,165 USD (3,06,15,677 INR) in type 2 SMA patients, and 40,735 USD (30,15,702 INR) in type 3 SMA patients. 12 Direct costs, including hospitalizations, special outpatient clinic visits, procedures/operations, and emergency care, accounted for a significant portion of this expenditure. Furthermore, costs for type 1 SMA patients increased more rapidly with age compared to type 2 and type 3 SMA patients. 12
Challenges in low-resource settings
The Patient Humanitarian Access Program, AVXS-101 program, compassionate use programs, and India's National Policy for Rare Diseases provide financial/medical assistance to SMA patients in accessing DMTs; the actual number of patients who obtain aid from these is minuscule. 5 Furthermore, a report by the Indian Organization for Rare Diseases highlights that there is a lack of coordination and delay in the availability of therapies for patients with rare diseases. Though 50 lakhs INR is provided by the government for the treatment of rare diseases, the overlapping of symptoms across rare diseases leads to delayed evaluations and longer processing times for approval of this aid. 56 It is important to note that early and continuous management of SMA without interruption is essential for achieving better clinical outcomes, particularly in the infant-onset type 1 SMA. It is crucial to adhere to the planned dosage schedule whenever feasible. 57 Based on the challenges observed in clinical practice by experts in the present study, it is likely that a loss of funding midway through treatment could lead to plateaus or even setbacks in improvement (Table 9).
Expert opinions on challenges in managing SMA.

CoE: Center of Excellence; NGO: Non-governmental organization; SMA: Spinal muscular atrophy.
The challenges in managing SMA are intensified by several factors, namely, (a) low prevalence of SMA; (b) limited awareness of SMA among the general population, HCPs, and policymakers; (c) inadequate expertise in diagnosing and treating SMA among HCPs; and (d) the chronic, degenerative, and life-threatening nature of the condition itself. All these factors collectively contribute to the complexities involved in effectively managing SMA.2,58
In diseases like SMA, where ongoing health and related services are necessary beyond a certain age, a smooth transition of care is crucial as patients move from childhood to adolescence and eventually to young adulthood. A few of the barriers highlighted by the MoHFW of the Government of India for having a transition of care include pediatricians hesitating to refer patients to non-pediatricians unfamiliar with childhood-onset conditions and doctors fearing loss of patient contact, which could have negative impacts on the patient's condition and follow-up care. In private settings, financial consequences may also be a significant reason. Patients and families may also struggle with the emotional transition from pediatric care, feeling anxious and fearing the loss of the pediatric care providers. Additionally, non-pediatricians may lack interest in pediatric diseases, perceiving them as additional responsibilities and the administrators may be unaware of the transitional care concepts, which may lead to reluctance to provide support without clear hospital policies. 59
The experts in the present study have highlighted various challenges related to affordability, accessibility, limited funding, inadequate referrals, and lack of awareness among HCPs regarding SMA in the Indian context (Table 9). Strategies for improving care and support for SMA patients in India as opined by the experts are presented in (Figure 2).

Expert opinions on future directions in managing SMA. DMT: Disease-modifying therapy; HCP: Healthcare practitioner; MDT: Multidisciplinary team; OPD: Outpatient department: SMA: Spinal muscular dystrophy.
Conclusion
SMA is a profoundly debilitating neuromuscular disorder that leads to the progressive loss of motor neurons in the spinal cord, resulting in severe muscle weakness and atrophy. SMA patients typically experience difficulties in basic motor functions, such as walking, sitting, and even breathing and swallowing. The burden of SMA is considerable in India with the current incidence rate being 1–2 per 1 lakh population with a carrier rate of 1:38. Unfortunately, there is a lack of awareness of SMA in India, due to which screening, diagnosis, and standard of care guidelines for this condition are also lacking. Most diagnoses of SMA are made only when the patient is >2 years of age as early detection of SMA is often missed due to the high mortality rates of those with type 1 SMA.
SMA management needs a multi-disciplinary approach along with DMTs as there is no cure for the disease. DMTs play a vital role in arresting the progression of SMA and reducing mortality and loss of motor functions.
Currently, Risdiplam is the only DCGI approved DMT with long-term data available on >250 patients in India and 16,000 patients globally; it is also the only DMT that can be orally administered with very few adverse reactions.
Key gaps in the management of SMA in India include the lack of standardized care guidelines and a lack of mechanisms to ensure patient access to therapy. Although the national rare disease policy is a promising step toward ensuring better management of rare conditions, it requires higher adoption rates and considerations for diseases such as SMA, which require lifelong high support interventions for effective management.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251386118 - Supplemental material for Spinal muscular atrophy in India: Patient journey, access to care, treatment barriers, and strategic recommendations: Insights from experts
Supplemental material, sj-docx-1-jnd-10.1177_22143602251386118 for Spinal muscular atrophy in India: Patient journey, access to care, treatment barriers, and strategic recommendations: Insights from experts by Sheffali Gulati, Anzeen Nazir Kanth, Ashwin Dalal, Monika Chhajed, Nehal Patel, Shrikant Jamdade, Surya Prabha, Vineeth Jaison, KN Vykunta Raju, Ajit Singh Baghel, Alpana Konderkar, Ann Agnes Mathew, Gurpreet Singh Kochar, Harsh Patel, Harshuti Shah, Ilin Kinimi, Jasodhara Chaudhury, AS Jyotsna, M Ranjith Kumar, Mary Iype, Mayank Nilay, Mayank Shrivastava, Neelu Desai, Praveen Kumar, Priyanka Madaan, Rahul Badheka, Rajni Farmania, Renu Suthar, Sakshi Ojha, Sanjukta De, Shikha Jain, Sidharth Shah, Smilu Mohan Lal, VY Vishnu and Aradhana Dwivedi in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
We would like to thank BioQuest Solutions Pvt Ltd for their editorial support.
Author contributions
All authors contributed to this manuscript as follows: Conceptualization, Writing – Original Draft, Writing – Review & Editing, and Formal Analysis. Additionally, Sheffali Gulati also contributed to Supervision and Visualization. All authors approved the final version of the article for publication and agreed to be accountable for all aspects of the work and resolved any issues related to its accuracy or integrity.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.