
Abstract
Nusinersen is a designer drug for spinal muscular atrophy (SMA) and was the first approved treatment for this once deadly disease. It is an antisense oligonucleotide that pairs with a specific locus of the Survival Motor Neuron 2 (SMN2) gene, to modify splicing and generate an increase in full-length SMN2 transcript. This in turn increases expression of survival motor neuron protein, deficiency of which results in motor neuron dysfunction and reduced cellular survival, the principal cause of SMA. Pre-clinical studies of nusinersen in animal models of SMA demonstrated substantial clinical responses and proof-of-concept, leading to successful clinical trials in symptomatic children and then in infants. Nusinersen's favorable safety profile after repeated lumbar intrathecal delivery as well as improvement in motor function and survival resulted in US regulatory approval for SMA in 2016. Other countries have followed with variable coverage policies depending upon age, weight, genotype and/or clinical severity. In the current treatment era, two populations of individuals with SMA exist: symptomatic patients identified in the clinic and pre-symptomatic patients (having no or few early clinical features of disease) largely identified by newborn screening. Real-world experience with nusinersen, the topic of this focused review, presents post-approval data in a broad range of patients beyond those studied in clinical trials. The favorable clinical response and safety profile are discussed, as well as the emerging new phenotypes of disease. Nusinersen, one of three FDA-approved drugs for SMA (as of 2025) remains an important therapeutic consideration for infants, children and adults with SMA.
Introduction
The spinal muscular atrophies (SMA) are a group of genetically defined disorders primarily affecting alpha-motor neurons in the brainstem and spinal cord, which lead to progressive skeletal muscle atrophy, weakness, and motor impairment. Classic SMA (OMIM 253300) was initially described by Werdnig and Hoffmann in the late nineteenth century. Over the next 7 decades three main phenotypes were identified and subsequently each has been identified to be due to homozygous deletions or mutations in the survival motor neuron 1 (SMN1) gene on chromosome 5q11.1–13.3, resulting in a lack of survival motor neuron (SMN) protein expression. 1 This protein is necessary for motor neuron development especially in the fetus and in early postnatal life, and subsequently throughout life for maintenance of motor neuron function.2,3 Humans have a silent “back-up” gene survival motor neuron 2 (SMN2) which differs from SMN1 in a few nucleotides, one of which (c.840C > T) in exon 7 affects a splice enhancer and which creates a splice silencer, thereby reducing inclusion of exon 7 into the messenger RNA. 4 This results in only a small amount of full length transcript being generated, estimated at approximately 10% per copy of SMN2 as compared to wild type. Humans vary in the number of copies of the SMN2 gene and this principally determines the amount of SMN protein generated, which in turn is inversely proportional, broadly speaking, to the severity of disease.5–7 While SMA has a broad spectrum of impairment with a continuous range in phenotype - from severe prenatal to more indolent adult onset - five clinical phenotypes have been constructed based mainly upon the age and features at presentation. 1 These types are useful as they can predict in a general sense the maximal level of motor function achieved and survival. In the current era of disease modifying treatments, phenotypes are shifting, but this overall classification scheme remains helpful. This is summarized in Table 1.
Classification of spinal muscular atrophy. Summary of SMA types with genotype associations, epidemiology, clinical features and survival for clinically manifesting individuals. This reflects a historical classification with supportive care, prior to availability of SMN-directed therapies. (with permission, adapted from Swaiman's Pediatric Neurology: Principles and Practice, 7th edition, “Classic Proximal Spinal Muscular Atrophy”, chapter 160, Elsevier, 2025).

Natural history
Approximately half of individuals born with SMA have the severe phenotype, type 1 (“Werdnig-Hoffmann disease”). These babies are born normal and develop onset of symptoms within the first six months of age (usually by 3 months), failing to gain independence sitting, and at risk for severe related morbidity due to bulbar and pulmonary dysfunction, leading to a median age of survival of approximately 1 year.8–10 Supportive measures such as gastrostomy tube placement for nutritional support and cough assist and non-invasive or invasive ventilation support has extended life expectancy for these babies, but unfortunately, in the absence of a disease modifying therapy (DMT) there is uniformly relentless progression of weakness to a quadriplegic state. 8 The majority of type 1 individuals have 2 copies of SMN2, but this alone is not sufficient to predict the phenotype. 6 Type 2 SMA (intermediate form, “Dubowitz disease”) occurs in approximately 25% of patients and is characterized by appearing normal at birth, developing the ability to sit and sometimes stand and bear weight but never gaining the ability to walk independently. 11 These children typically have a moderate degree of bulbar and pulmonary impairment requiring some degree of nutritional and ventilation support. They are prone to developing significant kyphoscoliosis that often requires surgical correction as well as joint contractures. With supportive care life expectancy is typically into the 3rd to 5th decade. 1 Type 3 SMA (Kugelberg-Welander disease) occurs in approximately 10% of patients and is characterized by normal appearance at birth and early infancy, gaining the ability to walk, but often loss of this skill as the disease progresses. 12 Diagnosis is typically in the 2nd to 4th year of life for the more severe form, type 3A, and up to age 18 or so for the milder form, type 3b. 12 Respiratory and musculoskeletal morbidities tend to be mild and life expectancy is normal. The two ends of the spectrum are the least common. Individuals with 1 copy of SMN2 nearly always have the most severe and rare phenotype (<1%), type 0, with severe weakness at birth, typically with joint contractures indicating lack of fetal movement, and with feeding and breathing compromise.13,14 Life expectancy is typically weeks. Individuals who have normal motor development and have symptom onset as adults are termed type 4. 15 They may develop slowly progressive weakness but are spared related morbidities and have a normal lifespan. 16
Scientific discovery, drug development and clinical trial readiness
The landscape of SMA changed dramatically after identification of the SMN1 and SMN2 genes in 1995 and subsequent characterization.17,18 This led to a rapid period of scientific exploration to understand the biology of SMA, develop transgenic animal models, and for clinicians to study genotype-phenotype relationships. This provided more discrete understanding of the natural history and the impact of standard of care support. The development of clinical outcome assessments and biomarkers proved useful in ensuing clinical trials. 1 Multiple gene targeted treatment strategies were explored, initially repurposed small molecule drugs which had positive in vitro response with increase in SMN protein expression but, unfortunately, failed to demonstrate any clinical response in clinical trials.19–22 Three novel gene targeted treatments for SMA were then developed and have been approved by regulatory authorities in many countries. The first of these disease modifying therapies (DMT) to be approved was an antisense oligonucleotide, nusinersen, delivered intrathecally on a periodic basis and which is the main topic of this manuscript.23,24 Risdiplam, a small molecule drug, is delivered orally daily and works in a similar fashion to nusinersen by modulating splicing of SMN2 to increase inclusion of exon 7 into the transcript and thereby increasing the amount of SMN protein in target motor neurons.25,26 Onasemnogene abeparvovec is a gene replacement therapy which delivers a normal copy of the SMN1 gene to motor neurons via a single intravenous delivery of an AAV9 vector.27,28
Clinical trials
Initial phase 1 and 2 studies for all three DMTs demonstrated safety and suggestions of significant efficacy. Successful phase 3 clinical trials conducted with these three DMTs in symptomatic individuals with SMA types 129–31 and 232,33 led to regulatory approval in many countries. Subsequently, there has been an additional focus to those identified shortly after birth, either by population based newborn screening (NBS) or from testing of an at risk individual due to a positive family history for SMA or from a positive carrier test in one or both parents.34,35 Clinical trials were conducted in “presymptomatic” individuals identified with SMA under 6 weeks of age having genetic confirmation but without definitive clinical features and/or markedly reduced motor compound motor action potential (CMAP) responses upon nerve conduction testing. In retrospect, some of the participants in these clinical trials, especially those with 2 copies of SMN2, demonstrated early features of disease at screening, e.g., hypotonia, reduced/absent reflexes, but were not excluded as these findings were not felt to be definitive. Additionally, the cut-off for CMAP eligibility was liberal, between 1 and 2 mV. All three DMTs demonstrated a favorable safety profile and even more robust clinical response when compared to the older symptomatic infants.36–39 While many individuals with 3 copies of SMN2 have demonstrated normal motor development in long-term extension studies, some beyond 5 years of age, those with 2 copies of SMN2 have in about half of the cases been delayed in the acquisition of motor milestones such as walking independently. 40 It is notable that nearly all of these individuals treated shortly after birth have thrived, with no mortality, few SMA-related hospitalizations for complications of disease, and a minority with 2 copies of SMN2 needing supplemental feeding or pulmonary support.
A long-term open-label extension study of individuals treated with nusinersen previously in clinical trials (NCT02594124, SHINE) has recently completed after 5-years of observation. Preliminary results reported at the 2025 Muscular Dystrophy Association meeting indicated a favorable long-term safety profile and one interim efficacy manuscript has shown sustained response in motor function – stabilization or improvement – especially in those without significant scoliosis. 41
It has become clear from these clinical trials that earlier treatment of the symptomatic patients with types 1 and 2 SMA do better with initiation of treatment soon after diagnosis and those treated shortly after birth with no or few features of disease do particularly well.
A recently completed study using a higher dose of nusinersen (NCT04089566, DEVOTE) has reported preliminary results with a favorable safety profile and efficacy, and may replace the current dosing schedule upon review by regulatory authorities. 42
More recently, a next generation ASO developed by Ionis Pharmaceuticals (ION306) came into clinical development by Biogen as salanersen (BIIB115) and is in a phase 1 clinical trial (NCT05575011) as of this writing. This newer ASO has the potential for longer duration of action and needing less frequent dosing.
The vast majority of newly identified patients are from newborn screening where this has been adopted for population screening. The commonly used DNA testing captures approximately 95% of the SMA population – those with biallelic deletions of SMN1 – and fails to capture the ∼5% with a deletion on one allele and a pathogenic variant on the other allele. This may change as testing strategies evolve, such as whole exome or genome sequencing. 43 While newborn screening is widely available in many parts of the world, including Europe, North America, parts of Asia, and Oceania, it has not yet been implemented in other large swaths of the world, including India and large parts of Africa and South America, leading to significant disparities in clinical outcomes. 44 Similarly, accessibility and criteria for accessing DMT can vary.
Symptomatic patients are in two categories: (1) those identified by newborn screening and treated as pre-symptomatic or with mild features of SMA, and (2) those identified in the clinic with fuller features of the disease and started on a DMT. Several general conclusions can be drawn from the clinical trials that are common to all three DMTs:
Response to these drugs is more pronounced in those treated shortly after symptomatic diagnosis than those with more advanced, chronic disease. Type 1 patients show a greater motor response than types 2 and 3 patients, suggesting that restoration of SMN in motor neurons is more impactful in infancy than later in childhood or in adults. The most robust response is noted in those identified and treated as neonates in a presymptomatic or mildly symptomatic state. Improvement in motor function in response to DMTs does not align with respiratory, bulbar or musculoskeletal issues. In general motor responses occur earlier and to a greater extent than what is observed in these other domains. Each of these DMTs has a favorable safety profile with individual benefits and risks:
Nusinersen requires repeated intrathecal administration and may have added risk from sedation for the procedure. Risdiplam is a daily oral medication, with less burden to administer than intrathecal drug delivery but where compliance may be an issue. Onasemnogene abeparvovec is a single administration and to date has demonstrated good durability of effect but also has a higher risk profile that requires more diligent and expert surveillance after dosing. New phenotypes have emerged in patients treated with these DMTs. There remains more to learn.
45
Systemic deficits and congenital abnormalities are seen in the more severely affected individuals with SMA. Severe type 0 patients are at a higher risk of congenital cardiac and vascular lesions.
14
Type 1 patients have metabolic abnormalities in mitochondrial and fatty acid metabolism.46,47 Other organ dysfunction (e.g., liver, kidney, pancreas) is uncommon in untreated and chronically treated individuals.
None of these DMTs are a cure and the question arises whether there is the capacity for further improvement by adding a second DMT. This has become more widespread in the US but is often limited by health insurance coverage policies. A phase 4 study sponsored by Biogen (NCT04488133, RESPOND) examines the safety and clinical and biomarker response to initiation of nusinersen following treatment with onasemnogene abeparvovec gene therapy, discussed more below. Other sequential and bridging strategies are being explored by clinicians on a less formal basis. 48 Some of this experience is captured in patient registries, as discussed in further detail below.49,50
In addition to these three SMN-focused DMTs other treatment strategies have emerged to address alternate targets or down-stream pathways. 51 These drugs are designed to be taken as an adjunct to a SMN-DMT, not as monotherapy. One strategy focuses on blocking myostatin, with the aim or promoting growth of muscle fibers and potentially increasing strength or endurance and related muscle function. 52 The most advanced clinical effort to date had promising results in a phase 2 study (NCT03921528, TOPAZ) and a phase 3 study (NCT05156320, SAPPHIRE) has recently completed with result pending. 53
Nusinersen
This review summarizes the post-marketing real-world experience of nusinersen, the first of three DMTs to receive regulatory approval in the US and Europe. This drug is an antisensense oligonucleotide (ASO), which uses the second generation 2′-O-2-methoxyethyl (2′-MOE) chemistry with an 18-nucleotide sequence and has a molecular weight of 7501 daltons. 54 This ASO binds via Watson-Crick pairing to a complementary splice silencer site in intron 7 near the exon/intron junction, displacing heterogeneous nuclear ribonucleoproteins (hnRNPs), thereby generating a more permissive environment in the pre-mRNA of SMN2 for inclusion of exon 7 and generates more of the full-length transcript and subsequently a greater expression of SMN protein. 55 Proof-of-concept was demonstrated in a Phase 2a study. 56 The current dosing regimen is 12 mg in 5 mL solution administered intrathecally, usually by lumbar puncture, in four loading doses in the initial two months, followed by maintenance doses every four months. 57 Class-specific ASO safety concerns are mainly for bone marrow and renal toxicity. Thus, pre-dose safety monitoring for prothrombin time (PT), activated partial thromboplastin (aPTT) time, platelet count and spot urine sample for quantitative protein assay is recommended. Of 346 individuals treated with nusinersen in the various clinical trials, the safety profile was favorable over several years observation. In the type 1 participants there was a slight increase in upper and lower respiratory tract congestion/infections, constipation, urinary tract infections as compared to the sham-controlled cohort. In the Type 2 patients there was a slight increase in pyrexia, headache, vomiting, back pain, epistaxis, falls, and respiratory tract congestion as compared to the sham control cohort. Some of these adverse events are likely attributable to a transient post-lumbar puncture syndrome or sedation related to the procedure. There may be a relative reduction in growth velocity. Rarely, aseptic meningitis, hyponatremia, and hydrocephalus have been reported, but are not clearly drug-related in all cases reported. In a small sample size of 6 participants in DEVOTE study, investigating a higher dose of nusinersen, rates of side effects attributable to LP were observed in 23% of injections, 42 comparable to the 21% at the approved dose of 12 mg. Anti-drug antibodies have been identified in approximately 6 percent of 294 participants receiving drug in these studies, sometimes transient and typically without related clinical symptoms or associated CSF abnormality. 58
Methods
This limited focused scoping review aimed to understand the real-world safety and efficacy experience with nusinersen in patients treated following marketing authorization and not participating in clinical trials. One biomedical database (PubMed) was searched using the terms “real-world” and “nusinersen” and “efficacy” between 19 April 2025–31 May 2025. Selected English-language studies of ≥5 patients describing treatment with nusinersen monotherapy were included in Table 2.
Summary of real-world evidence studies of nusinersen monotherapy.
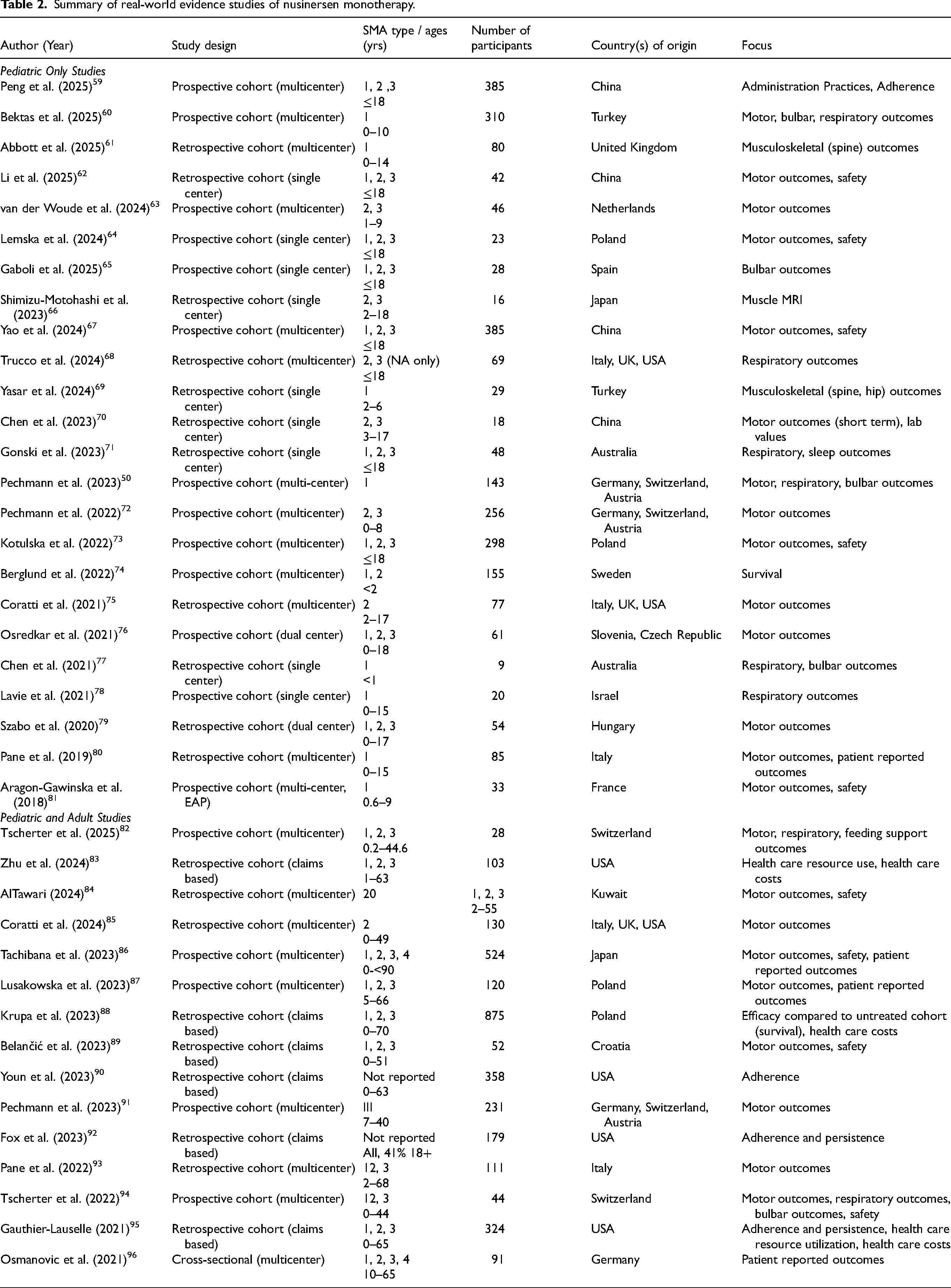
Studies were reviewed qualitatively, and information was extracted including geographic location, patient ages, patient SMA types, and focus of the study. As this article was not meant to be an exhaustive review, but rather, to capture the breadth and depth of the collective experience with nusinersen, some studies may have been missed and case reports with less that 5 participants were not included.
Results
Per Biogen, as of 2022, nusinersen was approved in 71 countries and over 14,000 patients have received this medication. 97 Overall, 55 studies were identified for inclusion. Of the studies identified in Table 2, 55% (n = 30) included patients from Europe, 18% (n = 10) from Asia/Middle East, 16% (n = 9) from North America, 4% from Oceania (n = 2), 2% (n = 1) from South America, and 5% (n = 3) were from Europe and North America. 31% (n = 17) were single-center cohorts, 55% (n = 30) were multicenter studies, 8 of which were from international consortia, 7 (13%) utilized claims data, and 1 (n = 2%) was a cross-sectional study. 44% (n = 24) of real-world evidence studies included only children, 35% (n = 19) included both children and adults, and 22% (n = 12) included only adults.
Postmarketing experience
Safety
When nusinersen was first approved by the FDA in 2016 in the United States for all ages and types of SMA, clinics had to grapple with ensuring safe and efficient administration to a broad range of patients. Dosing sites must ensure that appropriate multi-disciplinary care is available for the safe intrathecal administration of the medication. Teams must have designated staff members available to handle insurance authorization (if applicable) and must coordinate complex scheduling logistics to ensure patients receive scheduled doses according to the label.98,99 With such a broad initial approval in the United States, real-world evidence has been key to understanding the risk-benefit profile in those who were not studied in the pivotal clinical trials, and in identifying logistics hurdles that clinics may need to manage. In addition to ensuring systems are in place for efficient drug delivery, dosing sites should also ensure patients receive standard multi-disciplinary care.100–102 As benefits are more modest in those with significant symptoms of disease, standard of care is key in reducing morbidity and mortality from SMA. Institutional protocols, as well as multi-site registries, are needed to assess patient response and monitor disease trajectory over time. Such data is imperative in helping to define the new natural history of the disease in the current treatment era, and in monitoring outcomes to treatment. 45 While this review is focused on nusinersen, patients in some real-world settings are utilizing combination therapy. Thus, ongoing real-world monitoring for any emergence of unexpected side-effects in the setting of combination therapy will be critically important.
Patients with severe spine deformities were not included in initial trials. For such patients, dosing may be technically challenging and has necessitated alternative approaches. Real world techniques for administration to those with more complex spine anatomy have included ultrasound guidance by neurology providers, fluoroscopic or CT guided procedures with radiologists, cervical administration, or administration via a reservoir for repeated delivery.59,102–106 Use of sedation for lumbar puncture may vary based on age of the patient and geographical region. If sedation is being used, those with significant respiratory manifestations of disease may have increased risks from sedation, and so a careful assessment by a member of the sedation team should be performed prior to each dose. If illness symptoms are present, treatment should be postponed due to an increased risk of respiratory complications in the setting of illness.
Overall, nusinersen has demonstrated a well-tolerated safety profile. Safety events are summarized in Table 3, alongside some suggested management guidelines.
Reported side effects of nusinersen and suggested management considerations.

The label continues to suggest that clinicians check urine protein, platelet count, and coagulation studies at baseline and prior to each injection. 107 While platelet count and coagulation parameters must be monitored, there have been no reports of thrombocytopenia leading to clinically significant bleeding events. Similarly, while elevated urine protein is described in the clinical trials, there have been no reports of severe renal toxicity related to nusinersen.
Real-world side effects have mostly mimicked those described in the trials. Side effects related to LP are amongst the most common, and may include headache, nausea, vomiting, back pain, and in some cases, a more severe post-lumbar puncture syndrome (PLPS). Real-world studies have indicated rates of LP related side effects of 13–37%.67,86,87,108,109 PLPS has been reported at rates of 2–22% in both clinical trials and real-world cohorts.86,87,110 Rates in infants may be under-reported in studies, due to inability of these patients to report some of the clinical symptoms. In severe cases, this side effect may lead to treatment discontinuation.72,86,110,111 Management may include bed rest, fluids, analgesics, and anti-emetics. Rare cases of meningitis67,111–113 and hydrocephalus have also been described in patients who have received nusinersen. Notably, hydrocephalus was seen in untreated SMA patients at a fourfold higher rate when compared to unaffected controls, 114 suggesting that such observed cases may be related to the underlying disease state as opposed to a medication side effect. Nevertheless, clinicians should counsel patients on warning signs of hydrocephalus and obtain head imaging if there are clinical concerns. Like clinical trials, respiratory infections are commonly reported in real-world cohorts. 86 Notably, in one large adult cohort, respiratory infections were only seen in 5.4% of participants, a much lower rate than in the clinical trials. 111 This discrepancy reinforces the idea that the rates of infection seen in childhood trial participants are likely secondary to age and exposures and not due to the medication.
Efficacy
Clinical experience to date has broadly demonstrated benefit across the disease severity spectrum and has also added to the body of literature on the importance of early treatment. In a study of a broad age-range of 85 patients with SMA Type 1, the Italian Group showed a statistically significant improvement from baseline to 12 months on the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) in those under 5 years of age, and a statistically significant improvement on the Hammersmith Infant Neurological Examination — Part 2 (HINE-2) only in those <2 years. 80 Notably, of the 72 caregivers who completed questionnaires, 61 reported improvements, mostly attributed to motor function. 80 These results were similar in a broad-age range of 77 Type 2 patients from the International Spinal Muscular Atrophy Consortium (iSMAc), with positive mean changes in the Hammersmith Functional Motor Scale Expanded (HMFSE) and Revised Upper Limb Module (RULM) after 12 months of treatment when compared to natural history data. Similarly to the Type 1 patients, the largest predictors of response were age and baseline scores at treatment initiation, with those who initiated treatment at a younger age and those with higher scores responding more robustly. 75 In a large cohort of 385 symptomatic Chinese patients, improvements or stabilization on motor scales were seen across SMA types, although acquisition of new WHO motor milestones was very rare. 67 Similarly, in a large Japanese cohort of 584 patients, length of time since symptom onset was negatively correlated with treatment response across types of SMA. 86 The importance of early treatment has been critical in helping to expand access to newborn screening and thus, prompt initiation of presymptomatic treatment.
Those with severe neonatal onset Type 0 SMA were not included in clinical trials, and the decision to treat these patients necessitates ethics considerations. A handful of case reports describe the outcomes in these patients; most showed mild improvements on the CHOP-INTEND, with significant ongoing morbidity and/or morality.115–118 Additionally, patients with the need for continuous respiratory support and/or tracheostomy were also not studied. Real-world evidence has shown that even patients with more severe disease can have modest improvements on motor function scales, which may be clinically meaningful, although time since diagnosis remains the most significant predictor of motor benefit.
Treatment of babies with SMA before term gestation has been initiated in a small number of cases, either born prematurely and started on medication pre-term, or in one case the fetus was treated in utero. To date, these cases have had favorable safety and early features of efficacy.119–121 Given that in the early onset phenotypes, disease starts prior to birth, there has been marked interest in these early and prenatal treatment options. A recent paper outlined ASO delivery to the fetus in animal models via intra-amniotic injection and showed a significant increase in SMN protein expression in rodents and sheep without evidence of significant toxicity to either the fetus or mother. 122 The potential for such prenatal treatments is an exciting development and could lead to even more favorable outcomes given the importance of early treatment. However, such treatment offers unique safety challenges, and more information is needed. Nevertheless, the concept is an exciting prospect for the field.
Early clinical trials with nusinersen did not include adults, who represent an estimated 50% of patients with SMA in the United States, 123 and so real-world evidence helps define the risks, benefits, evolving phenotypes, and unmet needs in this population. Data from adult studies has overall shown benefit from treatment. One of the largest cohorts to date comes from a prospective observational study from the SMArtCARE registry, a multi-site consortium from Germany, Austria, and Switzerland. The study evaluated 237 adults. Using the HMFSE, RULM, and 6-min walk test (6MWT) approximately 30% of patients showed clinically meaningful improvements at 14 months, with 40% maintaining this level of improvement for ≥38 months. Another multi-center site studied compared 39 adults receiving nusinersen to 40 adults who were not receiving treatment. The nusinersen group had higher scores across multiple assessments, which was statistically significant at 16-months for the HMFSE, EK2, and 6MWT. Nusinersen treated patients showed higher rates of minimal clinically important difference (MCID) across all outcome measurements, but this was only statistically significant for the RULM, Egen Klassifikation 2 (EK2), and ALSFRS-R. On clinical reported and patient reported measures of change (CGI-C and PGI-G), 64.1% and 61.5%, respectively, reported an improvement to treatment, with a high degree of correlation. 124 Other large studies of adults have also demonstrated efficacy across ranges of adult ages and types108,109,124,125 Data utilizing patient reported outcomes generally indicate positive benefits from treatment,76,125,126 and one study reported improvements on quality of life measures when compared to baseline after 14 months on nusinersen treatment. 127
When compared to motor function, the benefit on respiratory and bulbar comorbidities of SMA has been more nuanced and subtle. In a study from the iSMAc group looking specifically at respiratory function in 69 children with Type 2 and non-ambulant Type 3 SMA, there was not a statistically significant in annual rate of decline of FVC when compared to untreated cohorts, although there was a trend treated patients reached a FVC threshold <60 at a later point when compared to untreated controls. Additionally, there was no change in non-invasive ventilation (NIV) requirements during the study period. 68 In another study of 48 pediatric patients in Australia, a small percentages of patients with Type 2 and Type 3 SMA were able to decrease their ventilatory support requirements, but these changes were not seen in Type 1 patients, and there was no change in the number of unplanned hospital or ICU days from respiratory illnesses. 71 Another study of symptomatic Australian infants showed that over half had a gastrotomy tube placed and initiated NIV during the study period, despite demonstrating modest motor improvements/stabilization. 77 These findings were similar in a Spanish cohort – all classic symptomatic Type 1 children had a gastrostomy tube placed by the end of the study. 65 In a large national registry of Japanese patients, only 1.7% of patients receiving ventilatory patients were able to discontinue that support. 86 Similarly, kyphosis and scoliosis may continue to progress even in those with more robust motor responses. 128 In one study from the United Kingdom of 80 Type 1 pediatric patients on nusinersen treated symptomatically, including some treated at a few months old, over 80% developed some type of spinal deformity. 61 Another Turkish study of 29 treated Type 1 patients showed that over 90% developed scoliosis and 60% developed hip subluxation. 69 There is ongoing discussion about the best approaches to manage such orthopedic complications, and the optimal time to intervene. There has been an increase in use of growth friendly devices to manage spinal deformities, 129 and as phenotypes continue to evolve, so too will interventional approaches. The discrepancy between motor, musculoskeletal, respiratory and bulbar responses may be due to tissue delivery, as nusinersen is typically administered via lumbar puncture. In autopsy data from five patients previously treated with nusinersen, higher drug concentration and protein expression was seen in the lumbar and thoracic regions of the spinal cord compared to the cervical cord. 3 In the DEVOTE study looking at higher doses of nusinersen, a larger percentage of those on the higher dose experienced stable or improved bulbar function at day 302, indicating that a higher dose may help mitigate some of this discrepancy. 130 However, longer-term data is needed, and additional focus on how to best improve these areas of ongoing need are critical.
Another area of ongoing interest and potential unmet needs is the cognitive development of those with SMA treated early in life. As most children with Type 1 SMA did not survive past the age of two, little was previously known on the cognitive profile of this group. Some early studies show that some of those with early onset SMA may be at risk of cognitive delays – two recent studies showed 35–55% of children showed at least some delays on standardized tests of cognitive developments. Factors associated with lower scores included male gender, two copies of SMN2, and use of permanent ventilation.131,132 Another recent study of those with Type 2 and Type 3 SMA found that intellectual disability was rare, and most patients had normal intelligence, and that some measures of cognitive performance may be impacted by motor function. 133 Questions have also been raised as to if there is an increased risk of autism in SMA. One small single center cohort reported rates of ASD of 21%, higher than the background rate of 5% seen in other patients in the state. 134 Efforts are underway to include more standardized assessments of the cognitive and neurodevelopmental domains into routine SMA clinic visits, but consensus is needed around optimal screening tools.
While newborn screening and early treatment of pre-symptomatic and pauci-symptomatic infants has drastically altered the phenotype of disease, some children, particularly those with 2 copies of SMN2, still develop symptoms of disease despite rapid treatment initiation. The Phase IV RESPOND study is looking at the addition of nusinersen in those who have suboptimal clinical status after receiving onasemnogene. 135 Participants were divided into two groups under 9 months and 9 months and over. Both groups showed an approximately 5-point improvement on the HINE-2 after 6 months of treatment. Serum neurofilament levels dropped by 70–80%. No serious AEs related to study drug occurred. 135 These results suggest that addition of a second agent may provide additional benefit in those at risk of more severe disease. As combination therapy is utilized differently - e.g., some use a bridge of an SMN2 modulator prior to use of onasemnogene abeparvovec, while others may continue an SMN2 modulator following treatment with this gene therapy – a proposed classification scheme for discussing combination therapy was developed. In the RESTORE registry (real-world study of onasemnogene abeparvovec), 5.6% were on combination therapy. 48 Understanding the new phenotypes for those who receive combination therapy is critical.
Rates of adherence in real-world cohorts have been variable. In a large cohort of 385 children in China, authors noted that over 96% of more than 2000 doses were received on time, with 81% of participants receiving all doses on time. 59 A retrospective adult study showed that over 90% of patients remained on nusinersen, and that approximately 70% of patients received all doses on time. 136 In contrast, a claims based study from the United States found adherence rates of approximately 70% for dosing schedule and found that only approximately 50% of patients who completed the loading dose phase remained on treatment at 12 months. Both non-adherent patients and patients who discontinued treatment had higher health care resource use (HCRU) and more medical complexity. 95 It is possible that patients in the United States may be more prone to delayed and missed doses given issues with insurance status and authorization, which is likely not seen in countries with nationalized health insurance systems. Given that with real-world administration, delays can occur for a variety of reasons, a helpful study looking at pharmacokinetics developed general guidelines on how to handle missed doses. If a dose is missed during the loading phase, resumption of dosing should occur as soon as possible. If a dose is missed during the maintenance phase, if the interval since the last dose is <4 months, the missed dose should be given as soon as possible with the next dose given according to the original schedule. If the delay is prolonged, loading doses may need to be given again. 137
Conclusions
Real-world evidence data supports a favorable long-term safety profile with repeated intrathecal administration of nusinersen in individuals with SMA. With up to 10 years’ experience, and thousands of patients treated to date across many countries, the depth and breadth of real-world evidence is significant. Clinical responses are consistent with observations from the earlier clinical trials and, broadly speaking, have demonstrated a sustained benefit in motor function (sometimes compromised by musculoskeletal complications) that is more substantial in patients identified pre-symptomatically and treated shortly after birth and in symptomatic patients treated shortly after clinical diagnosis. Improvement in bulbar and respiratory function tends to lag behind and is less substantial that that observed in skeletal muscle responses. Real-world evidence has been critical in helping to define the new and evolving phenotypes in the current DMT era. Gaps in understanding exist – notably, combination treatment remains an area where more research is needed. Current topics of interest include long-term cognitive outcomes and optimal management of the evolving musculoskeletal and bulbar complications, especially in the 2-copy population. While nusinersen is available in over 70 countries and thousands of patients have been positively impacted by treatment, major disparities in access continue to exist, with large swaths of the global population still unable to access treatment. Given the significant gaps in outcomes between those treated pre-symptomatically and those treated later in the disease course, continued advocacy around both newborn screening and prompt access to treatment is necessary to obtain the best possible outcome. Treatment with nusinersen continues to evolve, with a higher dose under consideration and with a next-generation molecule in clinical development. This drug, one of three currently (2025) approved for treatment of individuals with SMA, offers therapeutic benefit to individuals with SMA throughout the lifespan and with all degrees of severity of disease.
Footnotes
Acknowledgements
Funding support from Cure SMA to the PNCR SMA study group in which we participate is gratefully acknowledged.
Ethical considerations
This retrospective analysis of real-world evidence manuscripts did not require ethical committee review. No individual patient-facing data was included in this analysis.
Authors contributions
SEM and RSF jointly conceived and wrote this manuscript with equal contribution.
Funding
No external funding was utilized to support the preparation of this manuscript. Both authors are full-time salaried employees of non-profit health care institutions which enabled this academic activity.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Susan Matesanz has received personal compensation for advisory board/data safety monitoring board participation from Atamyo Therapeutics, Avidity Therapeutics, Novartis Gene Therapies, and Sarepta Therapeutics. She has participated as a site sub-investigator in Biogen-sponsored clinical trials and has also served as a site investigator in clinical trials for Dyne Therapeutics, Novartis, Pfizer, Roche/Genentech, and Sarepta Therapeutics. Richard Finkel has received personal compensation for advisory board/data safety monitoring board participation from AveXis, Astellas, Dyne, Italfarmaco, Novartis Gene Therapies, Inc./Novartis Pharma AG, Biogen, Catabasis, Ionis, ReveraGen, Roche/Genentech, Sarepta, and Scholar Rock; editorial fees from Elsevier for co-editing a neurology textbook; license fees from the Children's Hospital of Philadelphia; received research support from Biogen, Roche/Genentech; and participated as an investigator in clinical trials sponsored by Novartis Gene Therapies, Inc./Novartis Pharma AG, Biogen, Catabasis, Dyne, Genethon, Italfarmaco, ReveraGen, Roche/Genentech, Sarepta, and Scholar Rock.