
Abstract
Background and objectives
The severity of the phenotype of spinal muscular atrophy (SMA) is highly variable, yet little is known about the phenotypic variation among siblings. We systematically investigated the phenotypic variability of therapy-naïve 5q-SMA siblings leveraging a large multicentre cohort from the SMArtCARE registry.
Results
Clinical information was available from 132 siblings of 65 families. There were 24 (18.2%) type 1, 38 (28.7%) type 2, 54 (40.9%) type 3 patients, and 16 (12.1%) presymptomatic individuals. In 17 families (32.1%), there was discordance in the type of SMA among symptomatic siblings. We found no influence of gender on discordance in SMA type among siblings (p = 0.528). The median age at disease onset within all sibships varied by 6 months (interquartile range (IQR) = 1–30). There was no correlation in age of onset among siblings (r = 0.405; p = 0.052). Among siblings who lost ambulation, the median interval between the start of wheelchair use was 12 months, but the maximal interval was 18 years. In one pair of siblings, one sibling lost the ability to walk at the age of 13, whereas the other sibling was still ambulatory at the age of 54. In 6 sibling pairs (9.5%), only one of both siblings had a history of scoliosis surgery. Analysing SMN2 copy numbers, in one sibling pair (1.8%) 1 SMN2 gene copy was detected, while 10 (17.5%) had 2 copies, 23 (40.4%) had 3 copies, and 17 (29.8%) had 4 copies. Concordance in SMN2 copy numbers across siblings was observed in 90% of families. With increasing SMN2 copy number, the median differences in age of onset among siblings increased without reaching statistical significance.
Conclusion
This study reports considerable phenotypic variability in therapy-naïve SMA sibships that cannot solely be explained by differences in SMN2 copy numbers.
Introduction
5q-associated spinal muscular atrophy (SMA) is a monogenetic motor neuron disease caused by a biallelic defect in the SMN1 gene. Deficiency of SMN protein results in progressive loss of the lower motoneurons. 1 The incidence of SMA in Germany has been estimated at 1:7353. 2 The severity of the clinical phenotype in individuals carrying biallelic SMN1 deletions or variants varies widely, ranging from clinically asymptomatic individuals to severe atrophic tetraparesis requiring mechanical ventilation. 3
The most important variable influencing the severity of SMA is the SMN2 gene copy number.4,5 The SMN2 gene codes for a truncated SMN protein, expressing only about 5–10% full-length protein. 6 A higher number of SMN2 gene copies is usually associated with a later onset and milder course of SMA, although variations have been described.5,7–9
Detailed studies comparing siblings affected by SMA have been scarce. Jones and colleagues reported differences in SMA types in 15.2% of families. 10 Another study reported discordant siblings even in 60% of families, however, a more detailed classification of SMA types has been used. 11 A report of SMA siblings in terms of differences in clinical presentations, including age at onset and established motor scores is not yet available in larger patient groups, but case reports and smaller cohorts already indicated the occurrence of differences. Differences in the age of onset, spanning up to 15 years, were observed in a group of 101 siblings from 48 families, as reported in a study from 1994, with the diagnosis of 5q SMA confirmed in only 20 families through linkage analysis. 12 A case series showed differences in the age of onset of up to 24 years. 13 In 23 SMA families, 33 siblings were asymptomatic at an advanced age despite carrying biallelic SMN1 deletions.13–22
Since with nusinersen, onasemnogene abeparvovec xioi, and risdiplam three different disease-modifying treatments are now available, information about the expected differences in the severity of the disease among siblings is of great importance. This is underscored by the evidence of benefit from early or presymptomatic initiation of therapy in individuals genetically diagnosed with SMA identified by newborn screening programs.23–25 This study aims to describe the phenotypical differences of therapy-naïve siblings with 5q SMA in a large cohort of patients by analyzing achieved milestones, age at onset, SMN2 copy number, and established motor function scores, among others.
Methods
SMArtCARE is a disease-specific patient registry that has been collecting longitudinal data from individuals with genetically confirmed 5q-associated SMA from Germany, Austria, and Switzerland since 2017.
To analyze the sibling data, we considered the baseline data of all siblings registered in SMArtCARE as of December 2022. The data presented exclusively describe patients before the initiation of any disease-modifying treatments. Genetic data were collected within the registry including presence of a SMN1 deletion and SMN2 copy number. Examinations were performed in different laboratories. It is only documented if the SMN2 copy number measurement was performed by MLPA (multiplex ligation-dependent probe amplification) or PCR (polymerase chain reaction). For statistical analysis, we extracted data in terms of “year of birth”, “gender”, “age at symptom onset in months”, “sitting without support”, “walking without support”, “SMN2 copy number”, “RULM (Revised upper limb module) at enrolment”, “HFMSE (Hammersmith functional motor scale extended) at enrolment”, “scoliosis surgery”, “use of gastric or nasal feeding tube”, “type of ventilation”, and “wheelchair use”.
Since the data presented are the results of baseline measurements of therapy-naive patients, the SMA classification into severity levels 1–4 according to the German guideline “Spinal muscular atrophy, diagnosis and treatment” was used in this study. 26 The classification in the guideline is based on the age at onset of the disease and the motor milestones achieved, with the latter being decisive for the classification. Type 1 includes an age of onset up to six months and describes patients with pronounced proximal muscle weakness. If the age of onset is up to 15 months and the ability to sit freely is learned, they are classified as type 2. Type 3 describes patients with a later onset of the disease and if the ability to walk freely during the course of their lives is learned. If the onset of the disease is after the age of 30, it is classified as type 4. In a sub-analysis, siblings with SMA type 3 were divided into groups 3a and 3b, depending on whether the individuals were younger or older than three years of age at the time of diagnosis. Individuals were classified as presymptomatic if, despite genetic proof of 5q-associated SMA, no sign or clinical symptoms of SMA were apparent in an examination by a neuropediatric or neurological physician familiar with SMA.
Due to the real-world nature of the data, the complete data set was not available for each parameter studied. Concerning the SMN2 gene copy number, in the event of missing data or a discrepancy within the family, we contacted the responsible centers to obtain the copy number retrospectively. In the case of discrepant values, we arranged for a reevaluation through the Centre for Human Genetics in Cologne.
We performed statistics using SPSS (IBM SPSS Statistics, version 29.0.1.0). The chi-square test was used to calculate differences in the rate of discordance in SMA type between sibling pairs of the same and different sex. To describe the correlation of age of onset among siblings, we determined the intraclass correlation coefficient. To test for significant differences in age at disease onset as a function of SMN2 copy number, we performed a one-way analysis of variance (ANOVA). To calculate the statistical significance of differences in RULM or HFMSE in adult siblings, we used independent t-tests and Mann-Whitney-u tests.
Results
The study included 132 siblings with SMA out of a total of 1750 patients in the SMArtCARE registry at the time of data analysis. These were distributed among 65 families (63 sibling pairs (96.9%), plus two families with three siblings (3.1%)). The median age at baseline was 166 months (interquartile range (IQR)= 86–366). Out of the 132 individuals, there were 61 females (50.0%). In 15 (23.1%) sibships both siblings were female (including one family with three female siblings), in 20 (30.8%) families both siblings were male. In 30 (46.2%) families there were different genders (including one family with two male and one female sibling).
Looking at the distribution of SMA types across all 132 individuals, we found 24 (18.2%) type 1, 38 (28.7%) type 2, and 54 (40.9%) type 3 patients. There was no onset of symptoms in an individual after the age of 30 and thus no SMA type 4. In addition, 16 (12.1%) were presymptomatic at the time of baseline measurement (Table 1). The age at baseline measurement in these 16 individuals ranged from 0 to 21 months. Eight of these 16 presymptomatic individuals had older siblings with genetically confirmed SMA. The other eight presymptomatics are directly related within four families and at least one of each sibling was diagnosed by newborn screening. All of them were newborns or infants with an age ranging from 2 weeks to 21 months.
Baseline characteristics across all individuals. fam.=families. IQR = interquartile range.
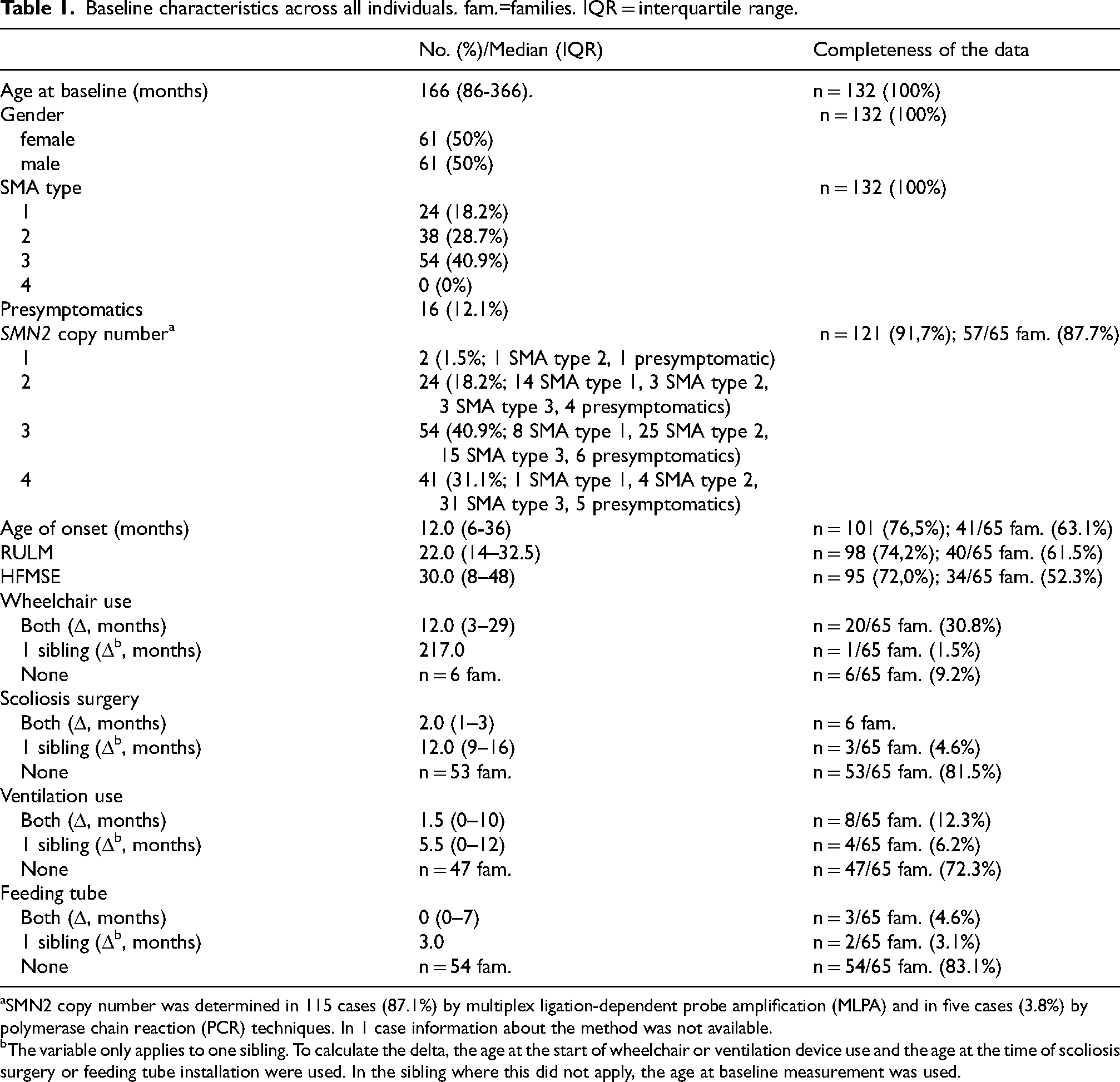
SMN2 copy number was determined in 115 cases (87.1%) by multiplex ligation-dependent probe amplification (MLPA) and in five cases (3.8%) by polymerase chain reaction (PCR) techniques. In 1 case information about the method was not available.
The variable only applies to one sibling. To calculate the delta, the age at the start of wheelchair or ventilation device use and the age at the time of scoliosis surgery or feeding tube installation were used. In the sibling where this did not apply, the age at baseline measurement was used.
Within the 53 sibships without presymptomatic individuals, discordance in SMA type was observed in 17 (32.1%) cases. The occurrence of SMA type 2 in one sibling and type 3 in the other was most frequently observed among discordant families (Figure 1). There was a gender match in 31 sibships (58.5%), correspondingly there were 22 sibships (41.5%) with different sexes. In 11 of 31 (35.5%) gender matched sibships and in 6 of 22 (27.3%) sibships with different genders there was discordance in SMA type. This distribution was not significantly different between the two groups in the chi-square test (p = 0.528). Specifically, discordance in SMA type was seen in 4 out of 12 cases (33.3%) in female sibships and in 7 out of 19 cases (36.8%) in male sibships. The difference in age at onset among same-sex sibships was a median of 6 (IQR = 0.25–24; n = 11) in female sibships and 6 (IQR = 1–24; n = 16) in male sibships. Within the 19 siblings with SMA type 3, the age at onset was available in 17 families to differentiate between SMA type 3a and 3b. In 4 cases (23.55%) a concordant SMA type 3a was found (once each female, three cases with different genders) and in 11 cases (64.75%) a concordant SMA type 3b (twice each female, four times each male, five cases with different genders). In 2 cases (11.8%) there was a discordance in SMA type 3 (once both male, once with a type 3a in the female sibling and 3b in the male sibling).
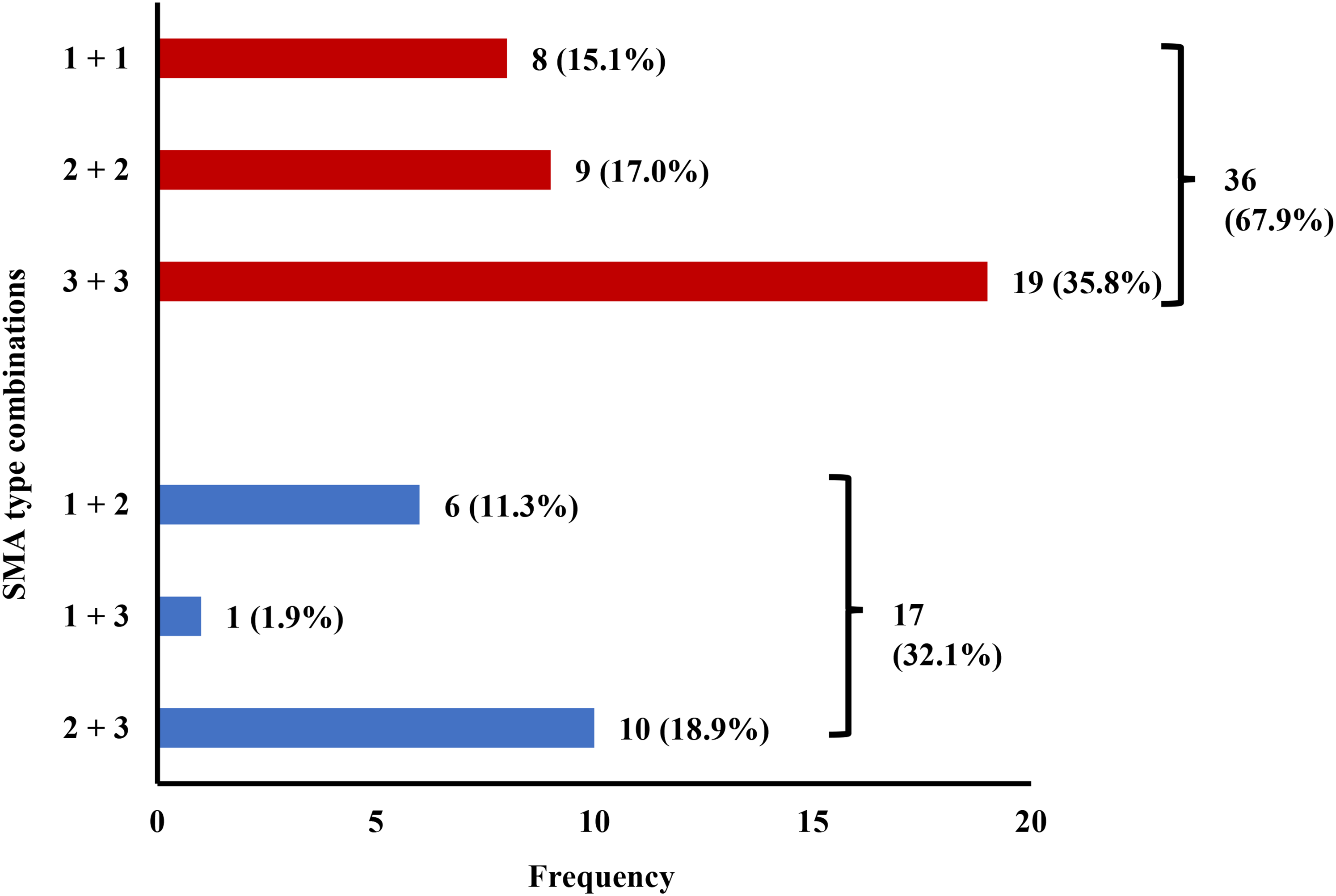
Combination of SMA types between symptomatic siblings (n = 53). Red columns represent concordant types, discordant types are marked in blue.
In 5 of the 6 families with different sexes and discordance in SMA type, a more severe phenotype was present in the male sibling. Data on age at onset were available in 16 sibships with different genders. The median difference in age at onset in this subgroup was 6 (IQR = 1–46.5). In 7 sibling pairs (43.8%), the onset of the disease was earlier in the female sibling, in 5 pairs in the male sibling (31.3%). In four cases (25.0%) it was identical.
Data on the onset of wheelchair use was available in 27 cases. The median difference was 12 months (IQR = 3–29). In one family, one individual was found to have started using a wheelchair at 65 months, whereas the sibling was still ambulatory at 282 months (23 years, 6 months). The median differences in the timing of scoliosis surgery were 2 months (IQR = 1–3), 1.5 months (IQR = 0–10) for ventilator use, and 0 months (IQR = 0–7) for gastric or nasal tube use (Table 1). In six families (9.5%) only one sibling required scoliosis surgery.
There was a median difference of 1 month among the siblings with regard to achieving the ability to sit or walk. In one family there was a difference in the loss of sitting ability of 132 months. In another family, an individual who was still able to sit was 76 months older at the time of the baseline measurement compared to the moment when his sibling lost this ability. Both these individuals showed 2 SMN2 gene copies. The median age of the latter concerning walking ability was 283 months. In particular, there was one family in which one sibling lost the ability to walk at the age of 13 years, whereas the other sibling was still ambulant at the age of 54 years. In this case, both siblings showed 4 SMN2 gene copies. In 6 families (9.2%), walking was learned by one individual, but never by its sibling.
The median age at onset across all individuals was 12 months, ranging from immediate perinatal diagnosis to 29 years (see Figure 2). The age at onset was available in all siblings in 41 families with a median onset of 14 months. There was a median difference in age at onset of 6 months (IQR = 1–30). The maximum difference in disease onset was 336 months (28 years). The individual with the later onset was male and showed 4 SMN2 gene copies, whereas his sibling with the earlier onset was female with no available SMN2 copy number. In 18 sibships (43.9%) the difference in age of onset was 12 months or greater. For sibling pairs with 2 SMN2 gene copies, respectively, median disease onset differed by 2 months, for sibling pairs with 3 SMN2 gene copies, the median difference was 4 months, and with 4 SMN2 gene copies, it was 24 months (Figure 3), however, these differences were not significant (p = 0.151).
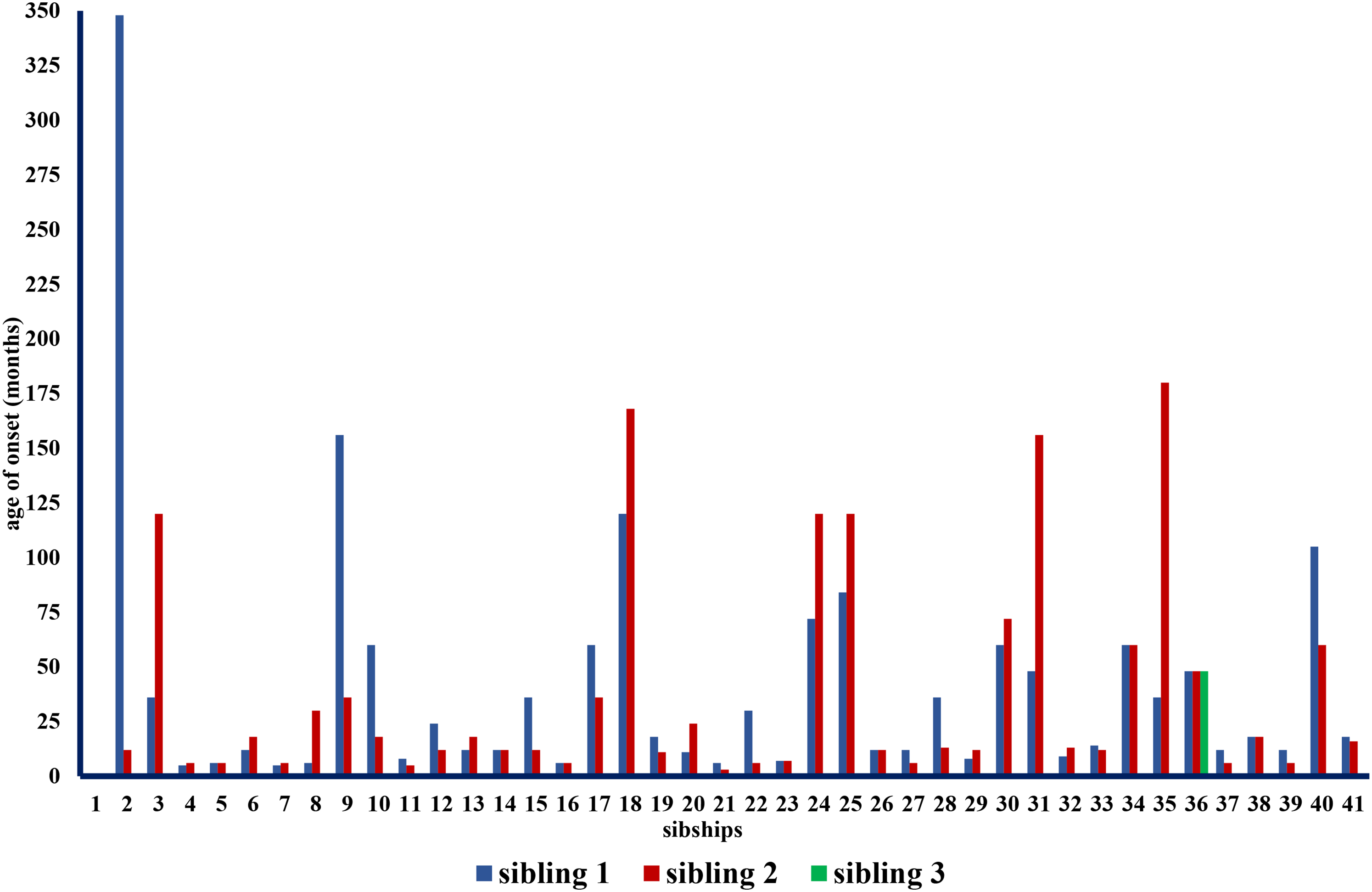
Age of onset in siblings from 41 families.
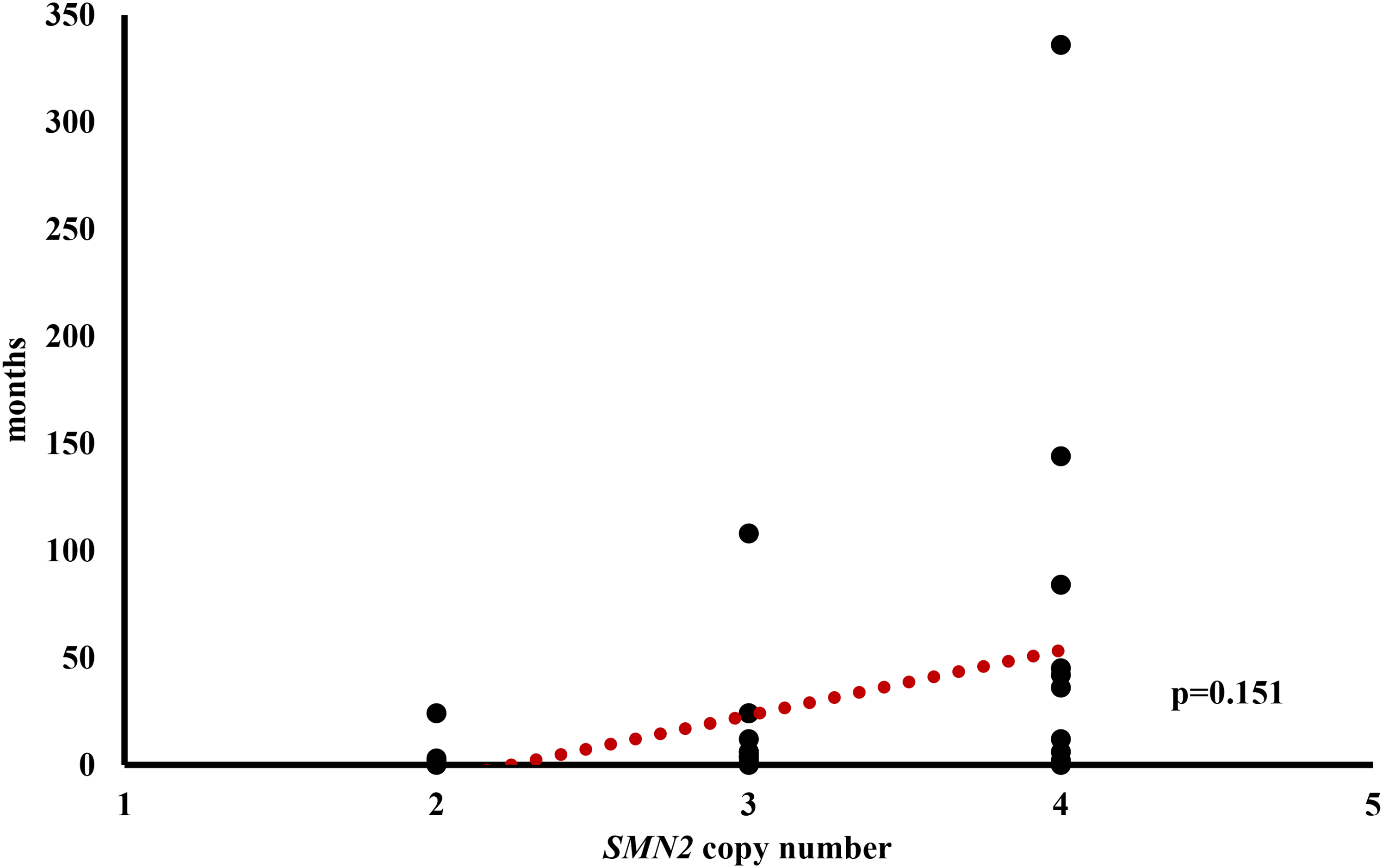
Differences in onset of disease in dependency of SMN2 copy number (n = 31).
To look at the correlation of age of onset between siblings across all families we determined the intraclass correlation coefficient. It showed no correlation (r = 0.425; p = 0.052).
We conducted a subanalysis of the data with regard to motor scores in families with adult siblings. Therefore, we looked at the sibling group with the higher scores in RULM and HFMSE and compared it with the group consisting of their more severely affected siblings. A median in RULM of 25 (IQR = 20–37) and a median in age of 31 years (372.6 months) were found among the adult SMA siblings (n = 19) in the mildly affected group. In the group of their more severely affected siblings, the median RULM was 20 (IQR = 11–25) and the median age was 31 years (370.9 months). The median difference in total scores was 7 (IQR = 3–14). The RULM scores differed significantly between the two groups (p = 0.032), but age did not (p = 0.483).
In the HFMSE (n = 16), the group of mildly affected siblings had a median score of 39 (IQR = 4–56) with a median age of 33 years (393.1 months). Their more severely affected siblings showed a median score of 7 (IQR = 1–25) and a median age of 32 years (379.9 months). The median difference in total scores was 18 (IQR = 2–30). There was a significant difference between the two groups in HFMSE score (p = 0.047), but not in age (p = 0.424).
SMN2 copy numbers were traceable from all siblings in 57 families. Matching numbers of SMN2 gene copies were present in 51 (89.5%) sibships. We found one sibship (1.8%) with 1 SMN2 gene copy each, ten (17.5%) with 2 copies each, 23 (40.4%) with 3 copies each, and 17 (29.8%) with 4 copies each. In six (10.5%) sibships there were discrepancies in SMN2 copy number. The most recent genetic report within these sibships was from 2018, in one case the SMN2 gene copy number was determined back in 2006. In two sibships with previously discrepant SMN2 gene copy numbers, re-determination at the Institute of Human Genetics at the University of Cologne revealed matching copy numbers. In the six sibships with discrepant SMN2 copy numbers, 4 cases had a matching SMA type. In one sibship, the sibling with 2 SMN2 copies showed SMA type 1 and the sibling with 3 SMN2 copies showed SMA type 3. In another family, the sibling with 2 SMN2 copies showed SMA type 3 with first symptoms occurring with 36 months and still able to walk at the age of almost 4 years at the time of data analysis, whereas the sibling with 3 SMN2 copies showed SMA type 2 with an age of onset with 13 months. In one family, the sibling with 3 SMN2 copies showed the first signs of the disease at the age of three, while the sibling with 4 SMN2 copies did not until the age of 14. Among the two sibling pairs with discordance in SMA type 3 (3a and 3b), 4 SMN2 gene copies each were found in one sibship. In the second sibship the SMN2 gene copy number was not available in one individual, and it was 4 in the other one.
Table 2 illustrates that 2 SMN2 gene copies occurred most often among the families with type 1 patients. In patients with SMA type 2, 3 SMN2 gene copy numbers dominate, whereas in type 3 patients there is a majority carrying 4 SMN2 gene copies. Within the families with discrepancies in the SMA subtype, 3 SMN2 gene copies were predominantly found in both the type 1 + 2 and type 2 + 3 combinations. There was no combination of type 1 and type 3 patients among the siblings with matching SMN2 gene copies. Among the 16 presymptomatic individuals one of them had 1 (6.3%) SMN2 gene copy, 4 (25%) had 2 copies, 6 (37.5%) had 3 copies and 5 (31.3%) had 4 copies.
Distribution of SMA subtypes within families with matching SMN2 copy numbers. Note that cases with presymptomatic family members were not included here.

Discussion
Our study reveals significant intrafamilial differences in 5q-SMA, as evidenced by variations in SMA type observed in more than one in three sibships. The discordance rate is higher than the results of Jones et al. who identified a rate of 15.2%. At the same time, it is significantly lower than the 60% found by Wadman and colleagues, although here a stronger subclassification of the SMA types was applied.10,11 If we combine the results of these studies with our data using the SMA classification into types 1–4 and solely directly related siblings, we obtain a discordance in SMA type in 72 out of 378 sibships (19.0%). If we look at the influence of gender on the discordance in the SMA type, we see a difference in the SMA type in 36 cases among the summarized 207 sibships with the same gender (17.4%). Among the 167 sibships with different sexes, we found 33 cases with discordance in SMA type (19.8%). It should be mentioned, that for the gender analysis only sibling pairs were considered from the study from Wadman and colleagues. Chi-square test showed no significant difference between both groups (p = 0.811). In summary, our data argue against a relevant influence of gender on the severity of SMA among siblings. In addition, our data provide a more detailed examination of intrafamilial variability.
When looking at the milestones achieved and lost, 23 (35.4%) sibships were found, in which both siblings were never able to walk. In 31 (47.7%) sibships both siblings are still able to sit, in 9 (13.8%) sibships both siblings are still able to walk. At the same time, however, on an individual level, there are repeated deviations with an emphasis on walking ability. Particularly striking are the three families in which one sibling lost the ability to walk and the second sibling is still ambulatory 16, 23, and 40 years later.
The median difference in age of onset among affected siblings is six months; however, there is again wide variation (Figure 2). It should be mentioned that this study included numerous infants or children at the time of their baseline measurement. The maximum difference in age of onset was 28 years. To our knowledge, the largest difference in age at disease onset reported previously was 24 years. 13 Regardless of asymptomatic biallelic carriers described in the literature, who may never develop the disease in the course of their lives, this data point helps to describe the range of possible ages of onset between siblings. In this particular family, the sibling with the later onset was male and at the time of baseline measurement (patient age 57 years) still ambulant, while his younger female sibling (age 55 years) lost walking ability at 42 years. The extent of these differences also becomes clear when looking at the intraclass correlation coefficient. The low r-value indicates that there is no correlation in the age at onset within the families due to repeated significant variations. 27
In a subanalysis we describe differences in established motor scores between adult siblings affected by SMA. We decided to look exclusively at adult SMA patients in terms of motor scores, as the natural course of SMA follows a much slower decline in motor skills as the patient reaches adulthood. 28 We contrasted the group of siblings with lower scores in RULM and HFMSE with the siblings with higher scores. There was no significant difference in age at the time of data collection in either motor score (p = 0.483 in RULM and 0.424 in HFMSE). When comparing the total scores however, there were significant differences in both RULM (p = 0.042) and HFMSE (p = 0.009). The median differences of 7 points in the RULM and 18 points in the HFMSE suggest relevance to everyday life and may, for example, determine in individual cases whether objects can be lifted and used or not. In one individual case, a 23-year-old patient presented a 20 points higher score in the RULM and a 57 points higher score in the HFMSE compared to his 27-year-old brother, while both patients showed 4 SMN2 copies. This underlines the large variance in the clinical phenotype of SMA. When assessing the motor scores, it must be considered that baseline measurements of HFMSE and RULM were not undertaken at the same age of the siblings. This is due to the retrospective approach of the study and the fact that an age-adjusted consideration of the motor scores would include a treatment bias, especially in the younger siblings. A closer look at the data reveals that in the RULM in 8 of the 19 adult families (42.1%) and in the HFMSE in 9 of the 16 adult families (56.3%) with a larger age difference, the older sibling showed better scores. The relevance of the age differences in the motor scores among the adult siblings can therefore be regarded as low overall.
Nevertheless, it should be emphasized that, especially concerning Figure 2, a large proportion of the siblings are similar in age at onset. Specifically, an identical age at disease onset was observed in 9 (15.8%) sibships, and in 22 (38.6%) cases the difference was a maximum of 6 months.
Numerically, the mean difference in age at onset within SMN2 concordant families increases with higher SMN2 gene copy number, although this does not reach statistical significance (Figure 3), possibly due to the low number of cases. Bearing this in mind, the data from our study have implications for genetic counseling. With the recent implementation of newborn screening for SMA in many countries, it is expected that more asymptomatic siblings with biallelic SMN1 deletions will be identified in the future. The data presented here allow for more precise statements regarding potential phenotypical differences or similarities among siblings. Clear recommendations exist for the treatment of newborns with 2 or 3 SMN2 gene copies, 29 but there is more debate about the best time for treatment initiation for individuals with 4 SMN2 gene copies. A recent analysis of 164 individuals with 5q-SMA and 4 SMN2 gene copies suggests a proactive treatment approach, noting an early onset in many of this subset of SMA patients. 30 However, if one child in the family already has SMA with 4 SMN2 gene copies, significant variations in the age of onset can occur in siblings. Therefore, deciding when to initiate therapy remains challenging. These data also have implications for genetic counseling of older asymptomatic siblings in which biallelic SMN1 deletions were detected, following a positive newborn screening result in a younger sibling. Considering the evidence for considerable variability in the age of onset among siblings and the risk of late manifestations of symptoms, genetic testing of older siblings should be performed, especially if the index case has 3 or more SMN2 copies.
A discordance of SMN2 gene copy number has been found in 10.5% of cases in our study cohort. This value is slightly higher than in comparable previous studies. 11 From a genetic point of view, it is conceivable that some of these discrepancies are due to unequal crossing over or gene conversion during meiosis. 31 In both sibships with discordant SMN2 gene copy numbers, which were re-determined at the Institute of Human Genetics in Cologne, matching copy numbers were ultimately found. Thus, it is reasonable to assume that most cases of discordance in SMN2 copy number are due to technically incorrect determinations in molecular genetic testing, as the complexity and error-proneness of the assay formerly used are widely known and modern MLPA (multiplex-ligation-dependent probe amplification) kits with higher accuracy emerged only a few years ago. 32 Our siblings were not always investigated in the same laboratory with the same method. Moreover, 6 out of 20 cases (30%) with 2 SMN2 copies suffered from SMA type 2 or 3, representing a higher proportion than expected as only 21% were reported in an earlier large study. 5 This can raise doubt on the correctness of SMN2 copy number determination.
A closer look at the 6 sibships with discordant SMN2 copy numbers revealed a difference in age at onset of 11 years in one family. At the same time, four of the 6 families showed an identical SMA type, and in one sibship one sibling had a more severe SMA type with a higher SMN2 copy number (SMA type 3 vs. 2 with 2 vs. 3 copies). Thus, doubts about the accuracy of the SMN2 copy number determination are also justified from a clinical perspective. Assuming genotypes regarding SMN1 deletions are identical in affected siblings and that in the vast majority of cases, there is a matching SMN2 gene copy, there must be other factors that influence the intrafamilial phenotypic variability of SMA.
A potential effect of gender on severity in SMA generally is suspected, with male sex being associated with a higher prevalence and severity of SMA.33,34 Although among the SMA type discordant sibling pairs in our cohort the male sibling had a more severe type more frequently (5 vs. 1 out of 17 cases), a similar pattern could not be seen when looking at the age at onset of the disease. Additionally, we also found large clinical variation within same sex sibships. The above-described siblings with differences in ambulatory status of 23 years were both men, whereas the couple with the difference of 40 years were both women. In the sibling pair with the difference of 16 years, the sibling who is still ambulatory is male. In this respect, the data correlate with the sibling data from the Cure-SMA study by Jones et al., which also showed no significant influence of gender on the severity of SMA among siblings.
The protective effect of altered regulation in the PLS3 or NCALD gene has been pointed out previously.22,35 In one specific sibship of our cohort showing 2 SMN2 copy numbers each but no SMA type 1 the variants c.859G > C and c.835–44A > G in SMN2 were ruled out previously. 36 However, epigenetic factors are also increasingly coming into focus. In particular, the status of methylation of certain DNA segments appears to influence the severity of SMA.37,38 In addition, several other factors influence the final amount and functionality of full-length SMN2 RNA and SMN protein: microRNAs, splicing factors, translation, degradation, and posttranslational modifications. 39 Identifying these variables would represent an important step for a better understanding the molecular pathogenesis and possibly even reveal new therapeutic options.
The real-world nature of this study with a considerable amount of data based on patient self-assessment or collected retrospectively, presents significant limitations. In milder affected individuals with advanced age, the onset of the disease is years, sometimes decades, in the past, which may result in variations in reported data. Furthermore, a single symptom marking the onset of the disease is not defined. Thus, obvious muscle weakness in a clinical test is indisputable as symptom onset, but subclinical signs such as fibrillations of the tongue, a tremor of the hands, or abnormalities in the EMG have also been described.16,17,40 The patient's self-perception, i.e., whether the motor abnormality is perceived as disturbing in daily life, could represent the difference between a subclinical sign and the first manifesting symptom of the disease. Serum biomarkers such as neurofilament light chain were identified in SMA 41 which might additionally indicate a prodromal stage of SMA, which previously has also been shown for amyotrophic lateral sclerosis. 42
This study provides only a cross-sectional analysis of clinical and genetic differences between therapy-naïve siblings with SMA, thereby precluding any conclusions about the varied effects of new therapy on disease progression.
Conclusions
In summary, our study provides the most detailed description reported so far of phenotypical differences among untreated siblings affected by 5q SMA. These differences cannot be solely attributed to variations in SMN2 copy number. Other factors such as SMN2-dependent modifiers influencing epigenetics, splicing, transcriptional, translational, post-translational modifications, or stability of the SMN-protein, as well as SMN-protein-independent genetic modifiers in other genes or environmental factors might be responsible. The age of onset of one sibling with SMA does not allow a reliable prediction of the age of onset in another sibling, especially in siblings with 4 SMN2 gene copies. The identification of further genetic or epigenetic factors, which, in addition to the SMN2 gene copy number, may determine the severity of SMA, is crucial and could even represent new therapeutic approaches in the future.
Footnotes
Acknowledgements
We would like to thank all patients and families that agreed to share their data within the SMArtCARE registry. We thank Bernhard Haller from the Institute of Artificial Intelligence and Medical Informatics (Technical University of Munich) for statistical advice.
SMArtCARE study group: Sabine Stein, Katharina Vill, Astrid Blaschek, Iris Hannibal, Birgit Warken-Madelung, Annika Roser-Unruh, Paul Lingor, Anne Buchberger, Ricarda von Heynitz, Petra Rau, Antonia Demleitner, Bernhard Haller, Christina Saier, Katharina Dörnbrack, Christine Mauz, Adrian Tassoni, Tim Kampowski, Franziska Wenzel, Sven Seilnacht, Sibylle Vogt, Manuel Pühringer, Marie-Luise Drax, Elisabeth Steiner, Sandra Bayer, Sandra Baumann, Johanna Feichtmayr, Nino Weiß, Viola Horneff, Kerstin Böcking, Sabine Hettrich, Sybille Stephan-Lutter, Christine Sprengart, Isabelle Meyers, Hanna Sophie Lapp, Maren Freigang, Colin Murphy, Sybille Döhler, Benjamin Stolte, Jaqueline Lipka, Melina Schlag, Svenja Brakemeier, Valerie Scherwietes, Andrea Hackemer, Lena Russ, Katharina Unverfehrt, Kyrikos Martakis, Hanna Küpper, Nadja Kaiser, Veronka Horber, Doris Roland-Schäfer, Eva Jansen, Esther Maihöfer, Birgit Meßmer, Deike Weiss, Jonas Denecke, Paula Steffens, Joenna Driemeyer, Sophie Fischer, Nora Töpper, Kathleen Weinreben, Andreas Hermann, Bettina Göricke, Tareq Mohamad Muhandes, Tobias Baum, Jörg Tiedemann, Ulrike Wolf, Mario Müller, Jasmin Bischofberger, Christina Knellwolf, Isabella Bischofberger, Adela Della-Marina, Andrea Gangfuß, Barbara Andres, Isabella Schreiner, Britta Holtkamp, Nina Rademacher, Anette Schwerin-Nagel, Joachim Zobel, Heidemarie Pilch, Bettina Behring, Stephanie Schüssler, Raphael Seebröker, Klaus Goldhahn, Katharina Müller-Kaempffer, Daniela Zeisler, Kathrin Bühner, Sabine Hermann, Sylke Nicolai, Simone Thiele, Eva Malm, Laura Grimm, Stephan Wenninger, Peter Reilich, Miriam Hiebeler, Wolfgang Wick, Sandra Schmich, Nicole Berberich, Marcel Mann-Richter, Heidi Rochau-Trumpp, Marion Schnurr, Guido Stocker.
ORCID iDs
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Biogen, Roche and Novartis provided financial support for the registry. Data collection and analysis was carried out by the academic SMArtCARE network independent of the commercial partner.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/orpublication of this article: B. Becker has nothing to declare; I. Cordts has nothing to declare; J. Becker has nothing to declare; R. Günther has received personal fees from Biogen and Hoffmann-La Roche and served on advisory boards from Biogen, Hoffmann-La Roche, ITF Pharma, Zambon and research support from Biogen, outside of the submitted work; M. Baumann received compensation for advisory boards and speaker honoraria for Novartis, Biogen and Roche; A. Eisenkölbl received compensation for advisory boards and speaker honoraria for Novartis, Biogen, Roche, and Sanofi; B. Fiedler has nothing to declare; M. Flotats-Bastardas has received consultant fees from Novartis, Roche and Biogen; M. Fleger has nothing to declare; T. Hagenacker has nothing to declare; A. Hahn has nothing to declare; E. Hobbiebrunken has nothing to declare; A. Bevot has nothing to declare; J. Jahnel has nothing to declare; J. Johannsen received compensation for advisory boards and funding for travel or speaker honoraria from Avexis/Novartis, Biogen, PTC, Pfizer, Roche, Santhera Pharmaceuticals and Sarepta Therapeutics; C. Kamm received honoraria as a speaker and advisory board member from Biogen, Roche and Ipsen and unrestricted travel grants from Merz; J.C. Koch has received personal fees from AbbVie, Biogen, Ipsen, Roche, Merz and Zambon; C. Köhler has nothing to declare; H. Kölbel has nothing to declare; W. Müller-Felber has nothing to declare; C. Neuwirth received honoraria for services for Biogen, Roche, Argenx, Sanofi and Mitsubishi Tanabe Pharma, unrelated to this work; B. Plecko has nothing to declare; M. Smitka has nothing to declare; A. v. Moers has nothing to declare; R. Trollmann received honoraria and personal lecture fees from Eisai, Desitin, Roche, Sanofi, and Biogen; M. Weiler has received advisory board and consultant honoraria from Biogen and Hoffmann-La Roche, and speaker honoraria and travel support for conference attendance from Biogen, outside of the present work, and is a member of the European Reference Network for Neuromuscular Diseases (ERN EURO-NMD); A. Ziegler has nothing to declare; S. Goldbach has nothing to declare; K. Probst-Schendzielorz has nothing to declare; H. Lochmüller receives support from the Canadian Institutes of Health Research (CIHR) for Foundation Grant FDN-167281 (Precision Health for Neuromuscular Diseases), Transnational Team Grant ERT-174211 (ProDGNE) and Network Grant OR2-189333 (NMD4C), from the Canada Foundation for Innovation (CFI-JELF 38412), the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279), the European Commission (Grant # 101080249) and the Canada Research Coordinating Committee New Frontiers in Research Fund (NFRFG-2022-00033) for SIMPATHIC, and from the Government of Canada Canada First Research Excellence Fund (CFREF) for the Brain-Heart Interconnectome (CFREF-2022-00007), and is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review; U. Schara-Schmidt has nothing to declare; M.C. Walter has received compensation for advisory boards and travel/speaker honoraria from Affinia, Argenx, Aro, Biogen, Edgewise, Pharnext, PTC, Roche, Santhera, and Sarepta; J. Kirschner JK has received honoraria for participating in advisory boards and/or symposia by Biogen, Novartis, Roche, and Scholar Rock. His institution receives funding for clinical research from Biohaven, Biogen, Novartis, Roche, and Scholar Rock; B. Wirth received travel expenses and speaker fees from Biogen and Avexis/Novartis; A. Pechmann has AP received compensations for training acitivities and grants from Biogen, Novartis and Roche; M. Deschauer received personal fees as a speaker/consultant from Biogen, Roche and Sanofi.
Data availability
All data included in this analysis are recorded in the SMArtCARE registry. Anonymized and aggregated data will be provided by the corresponding author upon request and approval of the SMArtCARE steering committee.