
Abstract
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder characterized by progressive muscle degeneration due to mutations in the dystrophin gene. In addition to the well-known musculoskeletal, cardiac, and respiratory symptoms, DMD also involves significant central nervous system (CNS) manifestations, including cognitive, behavioral, and emotional deficits that profoundly affect patient quality of life. Despite advances in RNA-based therapies targeting muscle symptoms, CNS manifestations remain largely untreated. Preclinical studies in animal models have shown promising results in addressing these deficits through CNS-targeted delivery of antisense oligonucleotides, highlighting the potential of intrathecal and next-generation systemic delivery methods. This review explores the latest advancements in RNA therapeutics for DMD, focusing on overcoming the challenges of CNS delivery to address both muscular and neurological symptoms.
Introduction
Duchenne muscular dystrophy (DMD) is the most common and severe form of muscular dystrophy in children, with an incidence estimated between 1 in 3500 to 1 in 5000 live male births. 1 It is caused by mutations in the DMD gene, resulting in the absence of functional dystrophin, a protein essential for maintaining the structural integrity and stability of muscle fibers. 2 The lack of dystrophin leads to progressive muscle degeneration, ultimately causing loss of ambulation, respiratory failure, and cardiomyopathy. Although traditionally seen as a muscle disease, DMD also significantly impacts the central nervous system (CNS). Cognitive impairments, learning disabilities, and neurobehavioral abnormalities, including autism spectrum traits and attention-deficit hyperactivity disorder (ADHD)-like symptoms3–8 are common in DMD patients, indicating a pressing need to address CNS pathologies in therapeutic approaches.
Recent years have witnessed a surge in RNA-based therapeutics to address muscle pathology in DMD, in particular through the development of exon-skipping approaches based on the use of antisense oligonucleotides (ASO). ASOs that promote exon skipping to restore the dystrophin reading frame have indeed shown promise, exemplified by the approval of several ASO drugs (eteplirsen, golodirsen, viltolarsen and casimersen) which has sparked further exploration into RNA-based interventions for DMD. 9 Other types of therapeutic approaches, such as gene replacement with microdystrophins or gene editing strategies, have also made tremendous progress in recent years. 9 However, while all these therapies primarily target muscle tissues, the CNS manifestations remain largely untreated, limiting improvements in quality of life. Addressing CNS comorbidities has thus emerged as a crucial frontier for DMD treatment.
This review will focus on the challenges and advancements in delivering RNA therapeutics to the CNS to treat neurobehavioral symptoms associated with DMD. Current strategies for CNS delivery include direct routes, such as intrathecal and intracerebroventricular (ICV) injections, and new developments of modified ASOs designed to cross the blood brain barrier (BBB) may offer possibilities for systemic approaches. We will examine DMD brain comorbidities, the existing mouse models with neurobehavioral deficits, and the preclinical evidence for CNS-targeted therapies. Additionally, we will discuss the potential benefits of postnatal dystrophin restoration in DMD mouse models and how next-generation ASO chemistries may enhance BBB penetration. By addressing the need for effective CNS-targeted therapies, this review highlights emerging opportunities in DMD treatment that hold promise for improving cognitive and behavioral health in patients. Encouraging results from animal models suggest that RNA-based therapeutics may eventually offer a more comprehensive treatment approach, though further research is essential to translate these findings to clinical outcomes.
CNS comorbidities in Duchenne muscular dystrophy
Beyond the well-known musculoskeletal, respiratory, and cardiac complications of DMD, cerebral involvement was already observed in 1868 by Duchenne de Boulogne, who reported that patients also exhibited below-average IQ, language impairments, and, in some cases, epilepsy. 10 More recent clinical studies have demonstrated the varied nature of these co-morbidities, ranging from cognitive disorders to emotional disorders. It is now estimated that up to 50% of DMD patients may be affected by these disorders.11,12 It has been observed that the nature and severity of these disorders vary from one individual to another. The presence of these cerebral co-morbidities and their inter-individual variability can be largely explained by the expression of several cerebral isoforms of dystrophin: Dp427, Dp140 and Dp71. In fact, the DMD gene has several alternative promoters which give rise to several isoforms (named according to their molecular weight), including the three isoforms mentioned above. The full-length Dp427 isoforms have three tissue-dependent alternative promoters named according to tissue specificity: muscular Dp427 (Dp427 m), cortical Dp427 (Dp427c) and Dp427 expressed in Purkinje cells (Dp427p). Depending on the location of the mutation, the number of dystrophins affected varies. Thus, a proximal mutation will affect only Dp427, and the more distal the mutation, the more it will also affect the shorter isoforms (Figure 1). Each of these isoforms is expressed at different stages and in different parts of the CNS. Dp427c is weakly expressed at the fetal stage of development but is strongly expressed after birth and is mainly present in forebrain and cerebellar neurons at post-synaptic levels.13,14 The same applies to Dp427p, which is not expressed during development, but in the adult cerebellum. 13 Dp140 is mostly expressed in early fetal brain, and in the cerebral cortex, hippocampus and amygdala in the adult brain. 13 Dp71 is highly expressed in the fetal stage and also after birth, and is ubiquitously localized in astrocytes in the CNS, with higher expression in the amygdala, hippocampus and cortex.13,14 Dp40 is also expressed in early neonatal stage until adulthood, and has a neuronal expression.15,16
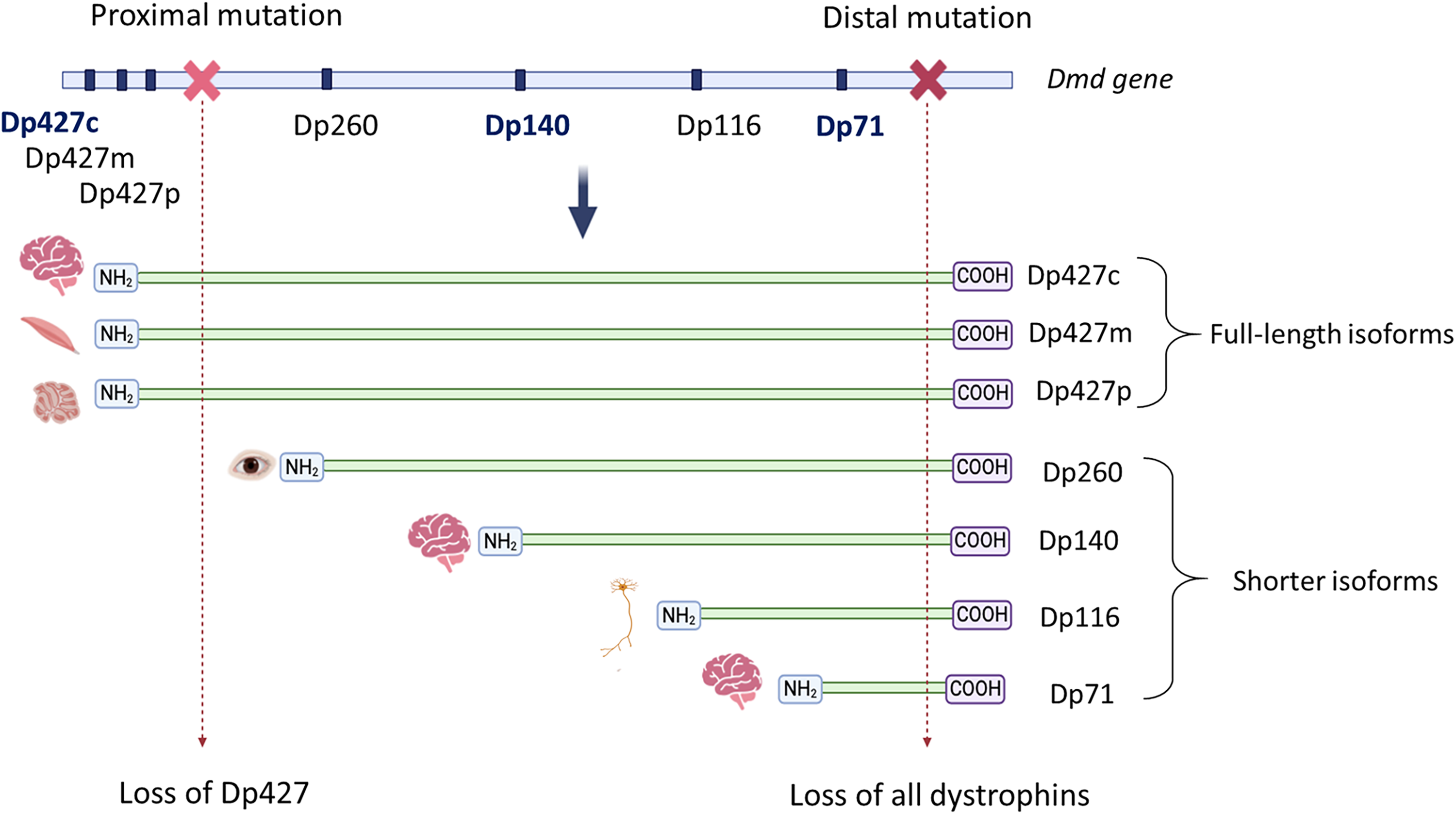
Visualization of a proximal or distal mutation in the Dmd gene and its consequences on the different isoforms. Representation of the Dmd gene with its various alternative promoters (dark blue bar), with the three brain promoters written in dark blue: Dp427c, Dp140 and Dp71. The gene products are shown below in green with their NH2 and COOH domains and their cell specificity: Dp427c (cortical), Dp427m (muscle), Dp427p (Purkinje cells), Dp260 (retina), Dp116 (Schwann cell), Dp71 (ubiquitous). The light and dark red crosses on the Dmd gene represent a proximal and distal mutation respectively, and their consequences on the gene product is represented by a dotted arrow.
To better understand the involvement of each isoform in these disorders, numerous studies have examined the correlation between genotype and phenotype in patients. Among cognitive disorders, it has been shown that the prevalence and severity of intellectual impairment increases significantly with the number of dystrophins affected.3,5,17–19 Patients with DMD show cognitive abilities about one standard deviation below the population mean, which remains stable over time without further decline. 20 A meta-analysis found that intellectual impairment prevalence is linked to cumulative dystrophin loss: 12% with Dp427 loss alone, 29% with combined Dp427 and Dp140 loss, and 84% when Dp427, Dp140, and Dp71 are all missing. 18 Verbal intelligence quotient (VIQ) is more affected than performance intelligence quotient (PIQ) in DMD, leading to academic challenges, particularly in reading and math, with these effects correlating with mutation locations.7,19 In addition to cognitive impairments, DMD patients often exhibit complex neurobehavioral issues such as anxiety, autism spectrum disorder (ASD), ADHD, and obsessive-compulsive disorder (OCD), with studies reporting a prevalence of 21% for ASD, 24% for hyperactivity, and 44% for inattention.3,6 The risk and severity of these disorders, particularly ADHD and anxiety, increase with the cumulative loss of Dp427, Dp140, and Dp71.4,17
Studies examining the role of dystrophin isoforms in the brain have highlighted their connections to various neurodevelopmental and psychiatric disorders. Research has demonstrated a functional association between DMD, and genes linked to conditions such as epilepsy, mental retardation, and cardiovascular issues.21,22 Specifically, the absence of Dp427 is linked to a decrease in GABAA receptors in the prefrontal cortex, suggesting its involvement in emotional disorders like anxiety due to impaired neuronal signalling. 23
Dp140, on the other hand, is associated with various early neurodevelopmental processes and its loss is correlated with an increased incidence of neurodevelopmental disorders.3,13 Meanwhile, studies in mouse models have shown that Dp71 is strongly associated with vascular development and interacts with molecules such as aquaporin-4 (AQP4) and Kir4.1, highlighting its role in cerebral vasculature and brain homeostasis.24–27 These findings collectively suggest that the loss of specific dystrophin isoforms contributes to both neurodevelopmental and vascular abnormalities in DMD.
Given the range of cognitive and behavioral challenges observed in DMD patients, the study of neurobehavioral phenotypes in DMD mouse models has been essential not only for understanding how dystrophin deficiency affects brain function but also for developing and evaluating potential therapeutic strategies targeting CNS comorbidities.
Neurobehavioral phenotypes of DMD mouse models
The most commonly used DMD animal model is the mdx mouse, which carries a spontaneous point mutation in exon 23 of the Dmd gene, leading to the loss of the Dp427 isoforms exclusively. 28 As the other isoforms have their promoters downstream of the mutation, their expression remains unaffected. In line with DMD patients’ symptoms, this mouse model also presents various cognitive and behavioral disorders. Mdx mice show memory impairments,29,30 particularly deficits in long-term memory, 31 along with various emotional disorders, such as increased anxiety and heightened fearfulness, which affect both unconditioned and conditioned defensive behaviors. 30 These memory disorders can be explained by abnormal synaptic transmission in the hippocampus where mdx mice exhibit abnormal long-term potentiation (LTP) mechanisms. This demonstrates the contribution of Dp427 to CA1 synaptic plasticity in the hippocampus.31,32 The alterations in GABAergic transmission found in patients are also found in mdx mice, with a reduction in GABAergic transmission due to reduced GABAA receptor clustering in the cerebellum, hippocampus, cortex and basolateral amygdala (BLA).30,33–35 Overall, findings from the mdx mouse model indicate that Dp427 plays a key role in stabilizing GABAA receptors, which may explain several of the observed symptoms.
To study the involvement of Dp140 in cerebral comorbidity, another dystrophic mouse model was used: the mdx52 mouse model, lacking exon 52 and therefore presenting cumulative loss of both Dp427 and Dp140. 36 Mdx52 mice exhibit more pronounced muscular and motor deficits than the mdx model, including reduced grip strength and enhanced musculoskeletal abnormalities, pointing to Dp140's role in coordination and higher centers of motor control. 37 The mdx52 mouse model exhibits more pronounced amygdala-dependent emotional and cognitive disturbances than the mdx model. Learning performance is significantly impaired with mdx52 mice showing delayed acquisition and retention in fear conditioning tasks, unrelated to differences in sensory perception or fear response. 38 Elevated anxiety and stress responses are also observed in behavioral tests such as the elevated plus maze and the open field and are likely linked to altered GABAergic signalling in BLA pyramidal neurons, which impacts excitation-inhibition balance. 32 This suggests a strong link between Dp140 and GABAergic transmission in the amygdala. Additionally, mdx52 mice display ASD-like asocial behaviors, attributed to reduced glutamatergic and altered GABAergic transmission in the BLA. 39
To study the cerebral role of Dp71, the Dp71-null mouse model was generated to allow selective loss of Dp71 expression without affecting other isoforms. 40 This model does not present motor disorders (no locomotion, coordination or equilibrium troubles), but does present cognitive disorders such as an altered strategy in goal-oriented navigation task. 41 This is likely due to alterations in glutamatergic transmission in the cerebellum, cortex and hippocampus. It was shown in the Dp71-null model that the cerebellum shows enhanced excitatory transmission and altered LTP in climbing fibers; 41 the prefrontal cortex demonstrates increased excitation, AMPA receptor disruption, and reduced LTP; 42 and the hippocampus displays disorganized presynaptic ultrastructure, with altered vesicle density and size. 43
Other existing mouse models can be useful to study cerebral comorbidities such as Dmd-null mice which present a deletion of the entire Dmd gene 44 or mdx3cv mice which express all dystrophin but at a very low level.45,46 It is important to note that certain brain disorders are also found in other dystrophic models such as the rat or the pig, and even the dog.47–50 This highlights the robustness of these symptoms across mammalian species, underscoring the value of animal models for evaluating potential brain-targeted therapies.
Delivery of ASO to the CNS of mouse models
RNA-based therapies, including ASOs for exon-skipping have achieved significant milestones in treating DMD, with some reaching regulatory approval in the U.S. and Japan. However, these therapies primarily target muscle pathology and do not address the cognitive and behavioral comorbidities linked to DMD, largely because of their inability to penetrate the BBB.51,52 Therefore, to investigate the therapeutic potential of ASOs on neurobehavioral comorbidities, it has become essential to explore local administration. Besides, in clinical settings, the intrathecal injection of an ASO (nusinersen) has been shown to be safe, effective and well tolerated after treatment of children affected by spinal muscular atrophy (SMA). 53
In fact, ASOs have been extensively administered directly into the CNS to explore their potential for treating neurological diseases in preclinical studies. The first clear evidence of ASO efficacy in the CNS was shown in 2006 when a gapmer ASO targeting SOD1 was administered to a rat model of amyotrophic lateral sclerosis, leading to reduced SOD1 mRNA levels and a marked slowing of disease progression. 54 This study catalysed the development of ASOs across various neurological disease models, with substantial phenotypic improvements documented in models of Huntington's disease, 55 SMA, 56 frontotemporal dementia, 57 Parkinson's disease, 58 prion diseases, 59 and several types of spinocerebellar ataxia.60,61 Most studies employed methoxyethyl (MOE)-gapmer ASOs, a widely adopted ASO design in clinical settings for neurological conditions.62,63 These studies have consistently demonstrated robust CNS distribution, ASO activity, and significant phenotypic prevention or reversal in these disease models. In larger preclinical species such as rats and non-human primates, intrathecal administration has been shown as the optimal delivery route to achieve widespread distribution and activity of MOE-ASO in the CNS.64–66 In mice however, ICV injection appears to be the most effective route. 66
In mouse models of DMD, Saoudi and colleagues have compared various delivery methods of ASOs to the CNS such as bilateral ICV, repeated ICV delivery using cannula implants in the lateral ventricles, continuous slow delivery with osmotic pumps and intra-cisterna magna injection (ICM) (Figure 2). 51 In this study, two different ASO chemistries were compared: the charge-neutral FDA-approved morpholino oligomer (PMO) and the lipid-conjugated tricycloDNA (tcDNA), currently evaluated in a phase 1/2a clinical trial (Avance 1 trial NCT05753462, sponsored by SQY Therapeutics). Bilateral ICV injection proved the most effective method for achieving broad ASO distribution and exon-skipping efficacy across CNS regions, although PMO yielded lower exon-skipping levels than tcDNA. Interestingly, the repeated ICV injections using cannulas did not significantly enhance exon-skipping efficacy for either ASO type. This was likely due to rapid ASO clearance when administered via cannula, as the continuous cerebrospinal fluid (CSF) flow under isoflurane anaesthesia (which does not slow the flow rate) could increase ASO turnover, thereby limiting the accumulation in the brain tissue. 67
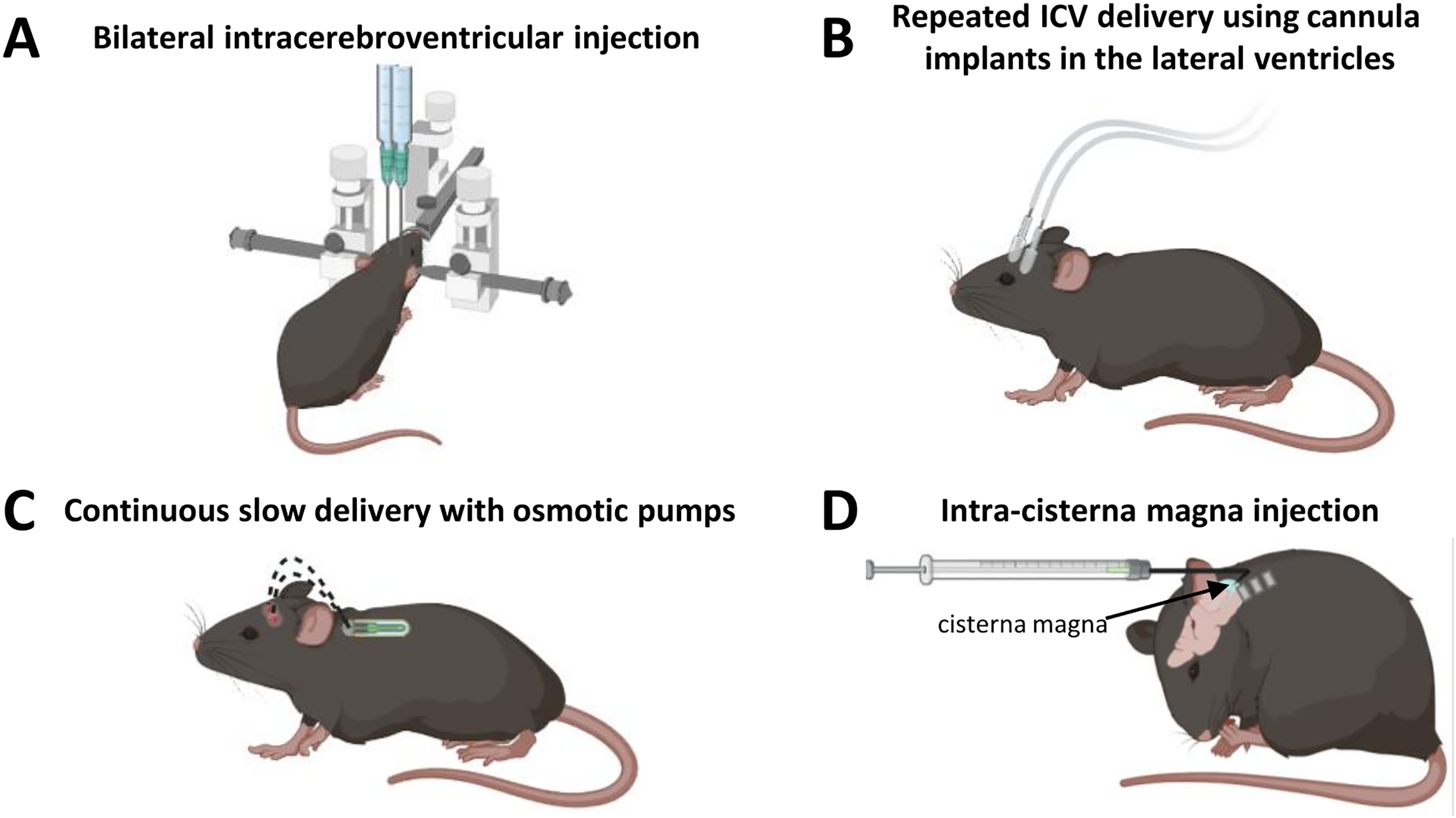
Summary of the different delivery routes of ASO to the CNS of mouse models. (A) ICV allowing a maximum of about 10 µl of ASO solution on deeply anesthetized mice. (B) Repeated ICV delivery using cannula implants in the lateral ventricles under isoflurane anesthesia. Infusions can be repeated between 3 and 5 times to reach maximum of about 50 µl of ASO solution. (C) Continuous slow delivery with osmotic pumps implanted under the back skin and directly linked to the ventricles via a cannula. The osmotic pumps can deliver a total volume of 100–200 µL of ASO solution through several days and weeks. (D) Intra-cisterna magna injections (ICM) of approximately 10 µL of ASO using a 30-gauge curved needle on deeply anesthetized mice. The ICM can also be combined with the bilateral ICV to inject a higher amount of ASO and to target a more widespread brain region.
Additional delivery techniques, including slow, continuous infusion using osmotic pumps and intra-cisterna magna injection, were also explored. The osmotic pump method allowed high ASO dosing, but did not improve exon-skipping levels, likely due to high CSF turnover in awake animals. These findings are in line with those from Rigo and colleagues 64 who had reported lower efficacy when using a very slow rate of infusion (0.5 µL/h), similar to the one of the osmotic pumps, in conscious animals. ICM delivery, which is minimally invasive and mimics intrathecal administration, led to ASO accumulation in the cerebellum and spinal cord, but limited distribution to other CNS areas, such as the hippocampus and cortex. Repeated ICM injections yielded somewhat better exon-skipping than a single dose but still fell short of the efficacy achieved with ICV. Overall, bilateral ICV injection of tcDNA-ASO appears to be the optimal method for inducing widespread CNS exon-skipping in mouse models of DMD. 51
Impact of postnatal restoration of dystrophin in the CNS of mouse models
The first studies investigating the impact of postnatal restoration of dystrophin in the brain were conducted in the mdx mouse model lacking Dp427. In mdx mice carrying a non-sense mutation in the Dmd exon 23, ASO are designed to skip exon 23 in order to restore the reading frame and the synthesis of a slightly shortened but functional Dp427. Sekiguchi and colleagues showed that direct ICV infusion of PMO targeting exon 23 results in partial restoration of brain Dp427 and improves the characteristic fear-motivated defensive behavior in these mice. 32 This was then confirmed using systemically administered tcDNAs which can cross the BBB at low levels and were shown to normalize this unconditioned fear response, despite the limited amount of Dp427 restoration.68,69 Further studies in mdx mice showed that it was possible to restore GABAA receptor clustering and normalize hippocampal synaptic plasticity through partial restoration of Dp427 by intra-hippocampal injection of an adeno-associated viral (AAV) vector encoding a U7-snRNA specific for exon 23 skipping. 70
More recently, Zarrouki and colleagues succeeded to rescue 5 to 30% of Dp427 in the CNS of mdx mice, after bilateral ICV of the palmitic acid conjugated-tcDNA-ASO containing a full phosphodiester backbone to skip exon 23 (tcDNA-Ex23).71,72 These restoration levels significantly reduced the abnormal unconditioned fear responses in mdx mice 7 weeks after ICV injection. Moreover, tcDNA-Ex23 restored long-term memory retention of mdx mice in an object recognition test but only slightly improved fear memory in the auditory-cued fear conditioning paradigm (Figure 3A). The partial restoration of the behavioral phenotype may be attributed to the limited expression and localization of Dp427 which was found in the synapses of the hippocampus and in the amygdala.
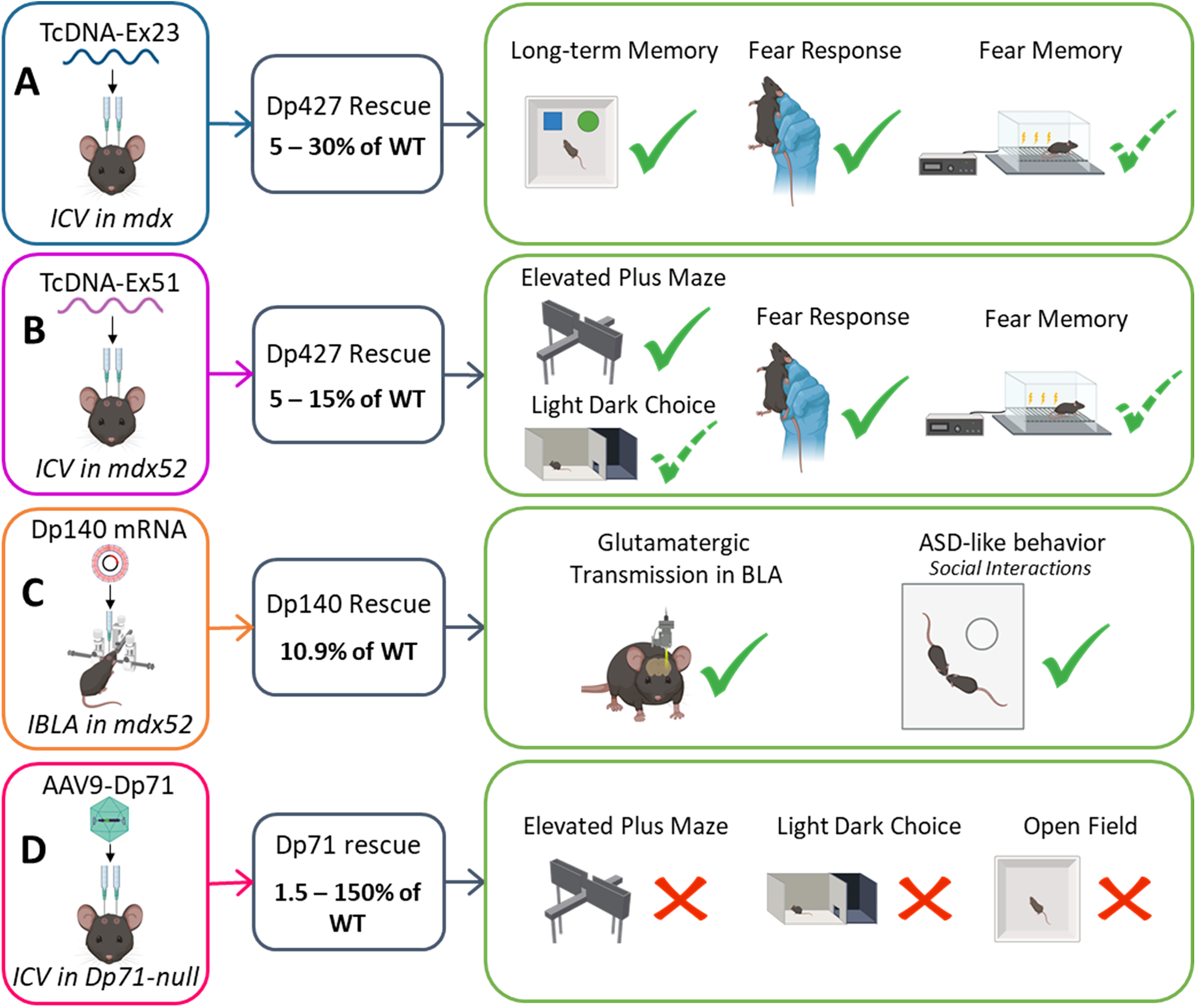
Representative selection of studies exploring the restoration of various dystrophin isoforms using different injection modalities. (A) Bilateral ICV of tcDNA-Ex23 restores 5 to 30% of Dp427 in cerebellum, cortex and hippocampus of mdx mice, and partially restores some behavioral outcome measures (adapted from Zarrouki et al., 2022). 71 (B) Bilateral ICV of tcDNA-restores 5 to 15% of Dp427 in cerebellum, cortex and hippocampus of mdx52 mice which fully or partially restore some behavioral outcome measures (adapted from Saoudi et al., 2023). 51 (C) Intra-basolateral amygdala (IBLA) injection of Dp140 mRNA-polyplex nanomicelles in mdx52 mice induces about 10% of Dp140 expression in the BLA and improve glutamatergic transmission in the BLA and autism spectrum disorders phenotype (adapted from Hashimoto et al., 2022). 39 (D) Bilateral ICV of AAV9-CBA-GFP-2A-Dp71 vector leads to the restoration of 1.5 to 150% of Dp71 expression in the cerebellum, cortex and hippocampus of Dp71-null mice but does not result in improvement of anxiety (adapted from Vacca et al., 2024). 73
Further experiments were recently performed in the mdx52 mouse, which is a particular relevant model carrying a deletion of exon 52, a region frequently found mutated in DMD patients. Furthermore, exon 52 deletion is amenable to two possible exon-skipping strategies that align with currently approved systemic therapies. Skipping exon 51 restores expression of the full-length Dp427 isoform but does not rescue Dp140, as its translation depends on the presence of the start codon in exon 51. In contrast, exon 53 skipping offers the possibility to restore both Dp427 and Dp140 isoforms. In this mdx52 mouse model, Saoudi et colleagues achieved between 5 to 15% of Dp427 after bilateral ICV injection of tcDNA-Ex51. 74 These levels of restoration were stable between 7 and 11 weeks and allowed to fully compensate the anxiety level measured in the elevated plus maze and partially reduce the anxiety in the light dark choice test. Moreover, in the unconditioned fear response test, partial Dp427 rescue significantly reduced the fear response although a large variability between animals was observed in this test. Finally, in the auditory-cued fear conditioning test, the treatment rescued the acquisition of the Pavlovian associative learning and partially improved the recall of fear memory which contrasts with the results of Zarrouki et al. where the partial rescue of Dp427 only slightly improved the fear learning and had no effect on the fear memory (Figure 3B). This difference may be explained by the more homogenous restoration of Dp427 across brain regions which may depend on each ASO specificity and also by the higher level of restoration in the cerebellum which can be implicated in fear-learning tasks. 75
Additional work in the mdx52 mouse model revealed that the lack of Dp140 was associated with abnormal social behaviors and decreased in glutamatergic transmission in mdx52 mice, 39 which are implicated in ASD-like symptoms in DMD patients lacking brain Dp140 isoform.3,76–78 Importantly, Hashimoto and colleagues demonstrated that these impairments in mdx52 mice could be improved using two distinct therapeutic approaches. First, they induced exon 53 skipping via repeated ICV injection of a PMO-ASO, resulting in approximately 1% expression of internally deleted Dp427 and 4.6% of internally deleted Dp140 in the BLA of treated mice compared to wild-type controls. To further investigate whether the observed improvements in glutamatergic transmission and social behavior were attributable to restoration of Dp140, Dp427, or both, the authors delivered Dp140 mRNA directly into the BLA of mdx52 mice using polyplex nanomicelles, thereby selectively restoring Dp140 expression. This strategy achieved approximately 10.9% Dp140 protein restoration without restoring Dp427 expression. Notably, this targeted rescue improved glutamatergic transmission and social behavior, although no significant effects were observed on fear response or anxiety-like behavior 39 (Figure 3C).
Studies were also conducted in the Dp71-null mice to explore the impact of postnatal restoration of Dp71. Dp71 is transcribed from a general promoter located in intron 62 of the dystrophin gene and can also give rise to a truncated isoform, Dp40. Its expression produces a 4.5-kb transcript that undergoes various alternative splicing events affecting exons 71 to 74, 78, and intron 77. These modifications result in three distinct isoform groups: Dp71d (including exon 78), Dp71f (lacking exon 78), and Dp71e (lacking exons 78–79 and containing part of intron 77). Among them, only Dp71d and Dp71f have been detected in the brain and retina, with Dp71d being predominant in the brain and Dp71f in the retina. Dp40, encoded by exons 63–70 (lacking exons 71 to 79), is also expressed in the brain and retina, although its function remains unclear. The localization of Dp71e is less well characterized. Finally, splicing of exons 71 to 74 results in several variants within the Dp71d and Dp71f groups, such as Dp71dΔ71 and Dp71fΔ74, following an established nomenclature. 79 In this context, Dp71d was expressed as a fusion protein (GFP-2A-Dp71d) from an AAV vector administered to Dp71-null mice either by systemic intracardiac injection in newborns, or by ICV injection in adults. Vacca and colleagues were able to rescue and re-localize Dp71d and some proteins associated with Dp71 (AQP4 and α1-syntrophin) in glial elements. However, the restoration levels were partial and insufficient to compensate the anxiety observed in Dp71-null mice (Figure 3D). 73 It was assumed that the Dp71d isoform likely does not contribute to anxiety regulation. Therefore, it was suggested that future attempts focus on restoring the Dp71f isoform, which accounts for one-third of the dystrophins in the brain and is likely to play a crucial role in regulating emotional phenotypes. 79
Altogether, these studies on postnatal re-expression of dystrophin in the brain have shed light on the role of each dystrophin isoforms but more importantly, they indicate that even partial restoration can alleviate some of the severe central comorbidities associated with DMD.
Challenges of CNS treatment in DMD
Despite these recent promising advances, several key questions remain: Can all behavioral deficits be improved, or even fully corrected? What is the optimal timing for intervention, would earlier restoration yield better outcomes? Additionally, the exact levels and brain regions required for effective recovery remain unclear. Targeted restoration of individual dystrophin isoforms in specific brain structures, such as the basolateral amygdala and cerebellum, could provide insights into each isoform's neurobehavioral roles.
Progress in this area is hindered by several challenges associated with achieving effective dystrophin restoration in the CNS. One major issue is the generally low level of protein recovery reported to date, which rarely exceeds 15%, despite exon skipping levels occasionally reaching 30–40%. This discrepancy between molecular and protein-level outcomes has been observed in several studies using the mdx52 mouse model and may be due, at least in part, to the 5′-3′ Dmd transcript imbalance which limits the pool of RNA available for effective exon 51 or 53 skipping.74,80 Another significant limitation lies in the delivery methods themselves. For example, in the study by Hashimoto and colleagues, eight ICV injections of PMO-ASOs were required to achieve only ∼1% Dp427 restoration. 39 To address this, the authors employed mRNA delivery using polyplex nanomicelles, which enabled local Dp140 restoration of up to 10.9% in the basolateral amygdala. However, this approach resulted in highly localized expression, insufficient for broad CNS coverage.
Additionally, some exons appear intrinsically more difficult to skip than others. For instance, effective exon 53 skipping in mdx52 mice required multiple ASO designs and formulations, yet still yielded only minimal Dp427 restoration (around 1%), as reported by Doisy et al. (2023). 81 This study also compared several ASO chemistries, including tcDNA, MOE, and PMO, and demonstrated that tcDNA-ASOs exhibit superior potency and broader exon-skipping activity across brain regions following ICV delivery.
Optimizing strategies to restore a broad and consistent expression of dystrophins across the CNS will likely be essential for effective intervention. Success in this area depends heavily on the potency of ASO drugs and the advancement of next-generation ASOs with improved CNS penetration and distribution.
Although ASOs can be administered directly to the CNS, as demonstrated safely and effectively with nusinersen in SMA patients, systemic delivery that reaches both muscle and CNS tissues would be particularly desirable for treating muscular dystrophies that impact both muscle and brain function. Toward this goal, extensive research is underway to develop ASO molecules with improved biodistribution properties and even capable of crossing the BBB.
Unlocking the CNS: the promise of next-generation ASOs
In the past few years, there has been tremendous progress in the development of new generation of ASO for the treatment of MDs. In particular recent advancements have focused on enhancing drug delivery to the muscle tissues, addressing the limitations of the approved “naked” PMOs with bioconjugation strategies. Among the most significant approaches, peptide-PMOs (PPMOs), which link cell-penetrating peptides to PMOs, have demonstrated improved cellular uptake and endosomal escape. For example, Sarepta's PPMO, vesleteplirsen (SRP-5051) had shown enhanced exon-skipping efficacy and dystrophin production in Phase 2 trials compared to eteplirsen (NCT04004065). Yet, Sarepta announced on November 6th 2024 their decision to discontinue development of SRP-5051 based on long-term safety and tolerability considerations (https://www.sarepta.com/community-letter-update-srp-5051-program). Another PPMO, PGN-EDO51 developed by PepGen to balance efficacy and safety, displayed initial success in healthy volunteers, and is currently in phase 2 trial in DMD patients (NCT06079736), although kidney toxicity remains a concern, with hypomagnesemia observed in some cases. Entrada Therapeutics is also developing PPMO conjugates based on cyclic peptides using an enhanced endosomal escape vehicle technology and has entered clinical trials. While these developments mostly aim at improving muscle uptake, some of these novel conjugates may be able to also target the CNS. Indeed, peptide conjugated PMO have previously been shown to target both peripheral as well as CNS tissues after systemic delivery in preclinical studies such as the PMO-internalizing peptide Pip6a in mouse models of SMA. 82 Other ASOs like tcDNA, have been shown to cross the BBB at low levels and rescue some behavioral features in DMD mouse models.68,72 More recently, Hasegawa and colleagues demonstrated that PMOs-based heteroduplex oligonucleotides were also able to induce low levels of exon skipping in the brain DMD mouse models, leading to an improvement of the unconditioned fear response. 83
Among these recent advancements, the development of antibody-oligonucleotide conjugates has drawn significant attention in the field and may offer promising opportunities to target the CNS. Avidity Biosciences and Dyne Therapeutics have developed ASO conjugates that both target the transferrin receptor 1 (TfR1) to improve muscle targeting, using either an antibody, or a fragment antibody, containing only the antigen-binding domain (Fab).84,85 Both platforms are currently under clinical evaluation in DMD patients and early data have shown potential for efficient muscle delivery, although clinical results have not yet been published in peer-reviewed publications and only communicated at conference and in press releases. Originally designed to enhance uptake in muscle and heart tissues, these innovative approaches also hold promise for penetrating the CNS, opening up new avenues for treating neurological aspects of muscular dystrophies. Indeed, TfR1 is highly expressed on brain endothelial cells comprising the BBB, which has sparked several groups to explore CNS drug delivery via TfR1-mediated transcytosis over the years.86–88 A recent study by Barker and colleagues demonstrated the remarkable potential of systemic ASO delivery targeting the TfR1. Their findings showed sustained knockdown of the targeted Malat1 RNA across various CNS regions and cell types in mice, including endothelial cells, neurons, astrocytes, microglia, and oligodendrocytes, along with skeletal and cardiac muscles. Notably, in cynomolgus macaques, the conjugated ASO targeting MALAT1 exhibited robust CNS delivery, achieving more uniform biodistribution and effective RNA knockdown compared to intrathecal administration of the same unconjugated ASO. These results highlight systemically delivered oligonucleotide therapeutics as a promising platform for crossing the BBB and achieving widespread CNS targeting. 89 While no published studies have yet demonstrated cognitive improvements following systemic treatment with a TfR1-targeted ASO in models of DMD, there is significant anticipation within the field as this approach could effectively address both peripheral and central deficits in muscular dystrophies.
Other strategies for delivering RNA therapeutics to the CNS have also been explored, including the use of extracellular vesicles (EVs). These naturally occurring EVs, released by cells, offer inherent protection to their cargo and have shown promise in efficiently and safely delivering therapeutics to the brain. EVs may represent a novel avenue for advancing RNA-based CNS therapies. However, further work is needed to optimize EV isolation, EV engineering for targeted delivery, cargo loading, and BBB penetration before they can be clinically evaluated. 90
Conclusion
Addressing CNS deficits in muscular dystrophies is essential for improving quality of life, as these symptoms profoundly impact cognitive, emotional, and social functioning. With longer life expectancies due to advancements in treating muscular symptoms, untreated CNS issues can severely limit patients’ independence and overall well-being. Developing effective therapies targeting both the peripheral and central symptoms is thus crucial for comprehensive patient care.
Preclinical studies in animal models have provided encouraging results, demonstrating that some neurobehavioral deficits are reversible following genetic or RNA-based interventions. Notably, intrathecal delivery of ASOs—already a standard treatment in SMA—could potentially be adapted to DMD patients to address cognitive symptoms. Such an approach may seem promising, but patient willingness to participate in trials raises questions, particularly given safety concerns. Direct CNS administration of charged ASOs has occasionally shown toxic effects in rodents91–93 and has infrequently been reported to induce ventricular enlargement in n-of-1 trials, underscoring the need for rigorous safety evaluations before clinical application.
Further research is also essential to clarify several key aspects about dystrophin restoration: the precise brain regions and dystrophin expression levels necessary for meaningful recovery, and how best to achieve consistent and broad CNS distribution. Optimizing ASO design, including next-generation ASOs with enhanced CNS penetration, is critical. The recent development of antibody-oligonucleotide conjugates holds promise as a dual-targeting solution for both peripheral and central deficits, potentially transforming the therapeutic landscape for DMD and other MDs.
Although challenges remain—ranging from improving targeting precision to ensuring long-term safety—these advancements bring hope. With sustained research efforts, the prospect of treating both peripheral and CNS symptoms of MDs may soon become a reality, offering patients a significantly improved quality of life.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Authors are supported by the Institut National de la santé et la recherche médicale (INSERM). OV and AG were also supported by the European Union's Horizon 2020 research and innovation program “Brain Involvement iN Dystrophinopathies” under grant agreement No 847826.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.