
Abstract
Classic phenotypes of plectinopathies include epidermolysis bullosa simplex and muscular dystrophy with proximal distribution, associated or not with skin blistering. However, in recent years, patients manifesting new muscular phenotypes including lower-limb distal weakness have been described. We aim to expand the phenotypic spectrum of plectinopathies by reporting a case presenting with a pure skeletal myopathy with predominant upper-limb distal weakness. We describe this distal myopathy phenotype providing comprehensive clinical, genetic, myopathological and radiological data. Genetic studies identified two truncating variants in PLEC.
Letter
Plectin, encoded by the PLEC gene consisting of 32 exons, is a large protein (>500 kDa) mostly expressed in skeletal muscle, brain and squamous epithelia. 1 Plectin is an intermediate filament-binding protein and plays a key role in the structural and functional organization of filamentous cytoskeletal networks, providing crucial biomechanical properties to tissues like skin or muscle.2,3 Eleven alternative first exons encode multiple plectin isoforms expressed in a wide variety of tissues, and, in particular, plectin isoforms 1, 1b, 1d, and 1f are highly expressed in skeletal muscle. 4 Plectinopathies comprise five autosomal recessive disorders, including epidermolysis bullosa simplex (EBS) which may associate with pyloric atresia (EBS-PA), muscular dystrophy (EBS-MD) and/or congenital myasthenic syndrome, limb-girdle muscular dystrophy type R17 (LGMDR17), and the autosomal dominant skin-only variant EBS Ogna. 5 Proximal shoulder and pelvic-girdle weakness is characteristic in LGMDR17,6–8 and predominantly proximal or scapuloperoneal weakness has been reported in EBS-MD phenotype. 9 Of note, plectinopathies are not considered within the group of distal myopathies.10,11
The last two exons of PLEC which encode the central rod and carboxyterminal domains are exceptionally large (approximately 3 and 6 kb, respectively). The EBS-MD phenotype is usually related with nonsense or frameshift mutations in exon 31.5,12,13 Despite new muscular phenotypes have been described in recent years with shoulder-girdle and lower-limb distal weakness,9,14 this is the first complete report of a pure skeletal myopathy with predominant hand involvement and no history of skin blistering.
An Asian 50-year-old man was referred to our clinic due to progressive distal weakness. He was born from unaffected unrelated parents. His father died from cardiovascular disease. He has a healthy maternal stepbrother. No family history of neuromuscular disease was reported. He presented around the age of 35 with slowly progressing intrinsic hand muscles weakness. Later, during the year before our first clinical evaluation, he also complained of frequent tripping due to anterior distal leg weakness. He never complained of sensory, ocular symptoms, fatigability nor skin lesions in the past. No bulbar or respiratory symptoms were reported.
First examination in 2022 showed slight strabismus without ophthalmoparesis, mild bilateral facial weakness, macroglossia with preserved tongue strength, moderate muscle atrophy of the anterior forearms and severe atrophy of the hand muscles, mostly affecting the interossei muscles and the thenar eminence (Figure 1(a)). No leg atrophy was evident on examination. Sensation was normal. There was symmetric distal weakness in upper limbs affecting finger extensors muscles (MRC 3/5) more than finger flexors (4/5), interossei muscles (3/5), abductor pollicis brevis (3/5), wrist flexion (4/5) and extension (3/5), and mild weakness in biceps and deltoid muscles (4/5). In lower limbs, strength was normal except for mild foot dorsiflexion (4+/5) and great toe extension (4/5) weakness. Deep tendon reflexes were abolished in upper limbs and normal in lower limbs. There was no scapular winging or joint contractures. He had no skin, dental nor nail abnormalities. No fatigable muscle weakness of ocular, bulbar or proximal limb muscles was elicited during physical examination.
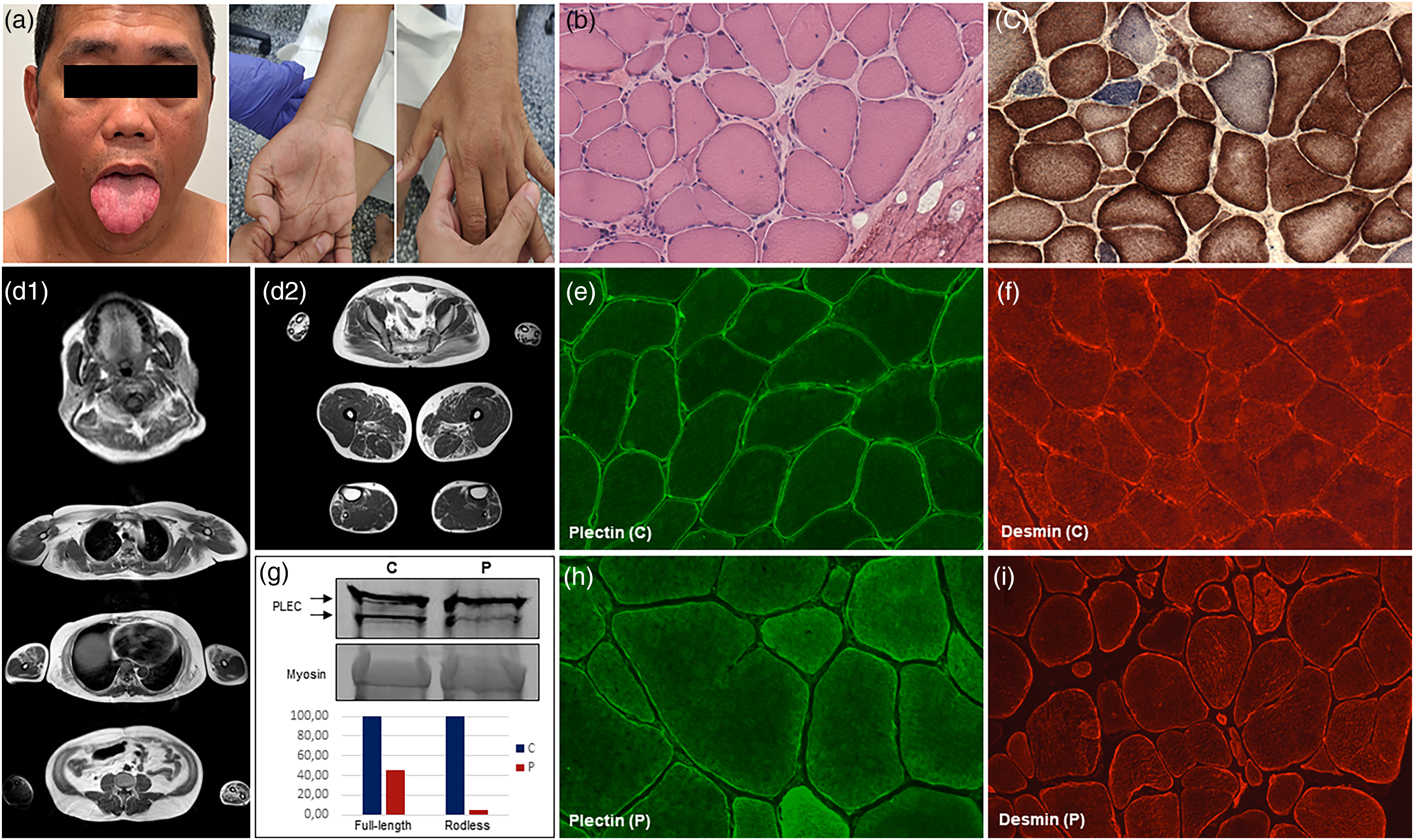
(a) image of the patient's tongue and hands. Macroglossia, severe intrinsic hand muscles atrophy, and mild thenar atrophy, are noted. Permission was granted by the patient. (b) Haematoxylin-eosin stain showed marked variability in fiber size, hypertrophic and atrophic myofibers, increased numbers of internal nuclei, fiber splitting, occasional necrotic fibers and increase of endomysial connective tissue. (c) SDH-COX reaction showed several COX-negative fibers. (d1) T1-weighted muscle MRI images of the tongue, shoulder girdle, upper limbs, and abdominal section. Note markedly increased fatty infiltration of the tongue, pectoralis major and minor, serratus, latissimus dorsi, and biceps brachii muscles, with symmetric involvement. Also note the severe muscle involvement of the anterior and posterior forearm compartments. (d2) T1-weighted MRI images of distal legs showing symmetric fatty infiltration of tibialis anterior and finger extensors muscles. At the thigh level, there was involvement of sartorius muscles and posterior compartments (biceps femoris short heads, semimembranosus, and adductors muscles). Pelvic imaging showed mild fatty infiltration of glutei maximus. (e, h) Plectin immunofluorescence (green, guinea pig anti-PLEC C-terminal Ab, Progen, Ref. GP21) in control (C) and patient (P) muscle, respectively, showed preserved plectin staining, but reduced intensity in patient muscle. Images are shown at 40X magnification. (f, i) Desmin immunofluorescence (red, anti-desmin Ab, Novocastra, ref. NCL-L-DES-DERII) showed subsarcolemmal and cytoplasmic desmin aggregates in patient muscle (20X magnification) compared to control muscle (40X magnification). (g) Western blot of muscle homogenates using plectin C-terminal antibody (GP-21, Progen) showed a markedly reduced amount of plectin in the patient (P) compared to the control (C). Densitometric analysis showed 45% and 5% expression of the full-length (upper band) and rodless (lower band) plectin isoforms, respectively, normalized to control levels. Myosin was used as a loading control.
CK levels ranged from 2600 to 4200 U/L. Nerve conduction studies showed slow amplitude of compound motor action potentials proportionate to the distal muscle atrophy in all tested motor nerves, except for posterior tibial nerves. This finding was more prominent in upper limb nerves (median and ulnar nerves). Motor and sensory nerve conduction velocities and latencies, and sensory nerve action potentials, were normal. Electromyography revealed occasional fibrillation potentials and positive sharp waves at rest and myopathic patterns in all examined muscles of the arms, hands and legs. Low-frequency repetitive nerve stimulation of right abductor pollicis brevis was normal.
Initial respiratory evaluation with spirometry revealed a restrictive pattern with a forced vital capacity (FVC) of 68% that decreased 16% on laying position, a maximal inspiratory pressure (MIP) of 63% and maximal expiratory pressure (MEP) of 30% predicted for age and height. A polysomnography showed a moderate sleep apnea syndrome. Cardiac evaluation including Holter and echocardiogram was normal.
Whole-body muscle MRI showed severe fatty replacement of the tongue (Figure 1(d1)). Also, severe muscle atrophy was observed in anterior and posterior forearm compartments and distal leg muscles, mainly involving tibialis anterior and finger extensors muscles. Mild involvement was observed at the pelvic girdle and thighs with a selective involvement of gluteus maximus, biceps femoris short head, semimembranosus, adductors and sartorius muscles (Figure 1(d2)). Of note, marked involvement of pectoralis, latissimus dorsi and serratus with preservation of paravertebral muscles, was noticed (Figure 1(d1)).
A muscle biopsy taken from the biceps brachii showed marked dystrophic features. Oxidative reactions showed some areas devoid of oxidative activity, abundant whorled fibers and several COX-negative fibers (Figure 1(b) and (c)). Sarcolemmal proteins were normally expressed except for mild decrease in dysferlin expression and utrophin overexpression at the membrane level. Immunofluorescence using two PLEC antibodies (N- and C-terminal) revealed preserved plectin immunostaining, with reduced intensity compared to control muscle (Figure 1(e) and (h)). Desmin immunofluorescence showed abnormal distribution with cytoplasmic and subsarcolemmal desmin accumulations (Figure 1(f) and (i)). Western blot revealed a markedly reduced plectin expression compared to control muscle, with no changes in the molecular weight of the protein (Figure 1(g)).
A custom gene panel (Nonacus, Birmingham, UK) including 139 myopathy-related genes identified two PLEC (NM_201378.4) nonsense variants: c.58G > T, p.Glu20* (exon 1f), previously reported in homozygous state in a patient with LGMDR17 7 ; and a novel variant c.5146C > T, p.Gln1716* (exon 31). Both variants, classified as pathogenic according to ACMG guidelines, 15 are absent from general population databases (gnomAD v4.1.0), and are predicted to be deleterious as loss of function is a known mechanism of disease in PLEC. Both the healthy mother and stepbrother were carriers of c.5146C > T variant in heterozygous state. Since no paternal sample was available, the phase of both variants was evaluated by Long Read Sequencing (using adaptive sampling of PLEC gene with Oxford Nanopore Technologies), confirming that both pathogenic variants are in trans. No other significant variants were identified in other genes linked to distal myopathies. Previous reports indicate that variants in exon 1f result in complete loss of plectin mRNA, 6 whereas truncating variants in exons 31 or 32 lead to a partial loss or reduction of plectin expression.3,9 In the case described here, as well as in previously published patients, the allele harboring the nonsense variant in exon 1f likely activates nonsense-mediated decay, preventing protein production. In contrast, the variant in exon 31 likely produces reduced levels of plectin isoforms, as observed in western blot (Figure 1(g)).
The clinical phenotype of our patient was characterized by the presence of muscle weakness and atrophy affecting predominantly the intrinsic hand muscles without skin lesions and, therefore, it cannot be classified as EBS-MD phenotype or LGMDR17. Also, supported by electrodiagnostic testing and physical examination, we could exclude a co-existing defect of neuromuscular transmission that can be associated with EBS-MD. Of interest, a family suffering from an adult-onset disto-proximal weakness with similar clinical characteristics including severe weakness in distal upper-limb muscles, mild facial weakness and strabismus, in combination with cardiac involvement, has been briefly reported in a scientific communication. 16 We consider important to highlight the highly increased blood CK levels that presented our patient, like most muscle phenotype reports of plectinopathies (ranges from 3.000 U/L to 4.000 U/L). Since this feature is rare in distal myopathies, differential diagnosis requires to consider Miyoshi myopathy due to DYSF or ANO5 variants, although these entities predominantly involve calf muscles. On the other hand, patients with mutations in other genes associated with distal upper-limb weakness, such as FLNC and TIA1, usually present lower CK levels.
To conclude, we aim to further expand the knowledge of this plectin-related predominantly distal myopathy phenotype providing detailed characterization of an affected individual. This observation contributes to broadening the phenotypic spectrum of plectinopathies, and we recommend considering plectinopathy in the differential diagnosis of distal myopathies, particularly when there is significant involvement of the hand muscles.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Instituto de Salud Carlos III co-funded by ERDF/FEDER (Una manera de hacer Europa) under grant FIS PI21/01621 awarded to M.O and grant FIS PI22/01859, SGR-Cat 2021 (AGAUR-Generalitat de Catalunya) through the project “Genomic medicine and rare diseases group” (2021-SGR-00835, 2023–2025, A.S.C, P.G, L.G.Q), and “Fundación Isabel Gemio”. LL is supported by a Grífols-funded grant (Beca de formación en enfermedades neuromusculares Isabel Illa). A.S.C. was supported by the Ministerio de Universidades (Spain) with grant FPU20/06692.
L.L, A.S.G, E.G, P.G, L.G.Q, and M.O are members of the European Reference Network for Neuromuscular Diseases.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.