
Abstract
The continued evolution of genomic technologies over the past few decades has revolutionized the field of neurogenetics, offering profound insights into the genetic underpinnings of neurological disorders. Identification of causal genes for numerous monogenic neurological conditions has informed key aspects of disease mechanisms and facilitated research into critical proteins and molecular pathways, laying the groundwork for therapeutic interventions. However, the question remains: has this transformative trend reached its zenith? In this review, we suggest that despite significant strides in genome sequencing and advanced computational analyses, there is still ample room for methodological refinement. We anticipate further major genetic breakthroughs corresponding with the increased use of long-read genomes, variant calling software, AI tools, and data aggregation databases. Genetic progress has historically been driven by technological advancements from the commercial sector, which are developed in response to academic research needs, creating a continuous cycle of innovation and discovery. This review explores the potential of genomic technologies to address the challenges of neurogenetic disorders. By outlining both established and modern resources, we aim to emphasize the importance of genetic technologies as we enter an era poised for discoveries.
History of genomic technologies in neurogenetics
Neurogenetics, at its core, aims to understand the link between genes, behavior, the brain, and neurological disorders and diseases. 1 Its origins trace back to the late nineteenth century when clinical descriptions of neurological traits and diseases laid the groundwork for modern nosology. Early observations of familial inheritance patterns foreshadowed the genetic underpinnings of diseases. In 1863, before the word “heredity” was coined, Nikolaus Friedreich described a form of juvenile-onset ataxia in children from families with seemingly unaffected parents. Today, we recognize this as Friedrich's ataxia, the most common form of hereditary ataxia, caused by biallelic GAA repeat expansions in the FXN gene.2,3 In 1886, Jean-Martin Charcot, Pierre Marie, and Henry Tooth described a spectrum of peripheral nerve disorders. Today, Charcot-Marie-Tooth (CMT) disease is the most common inherited neuromuscular disorder and has been associated to mutations in over 100 genes. 4
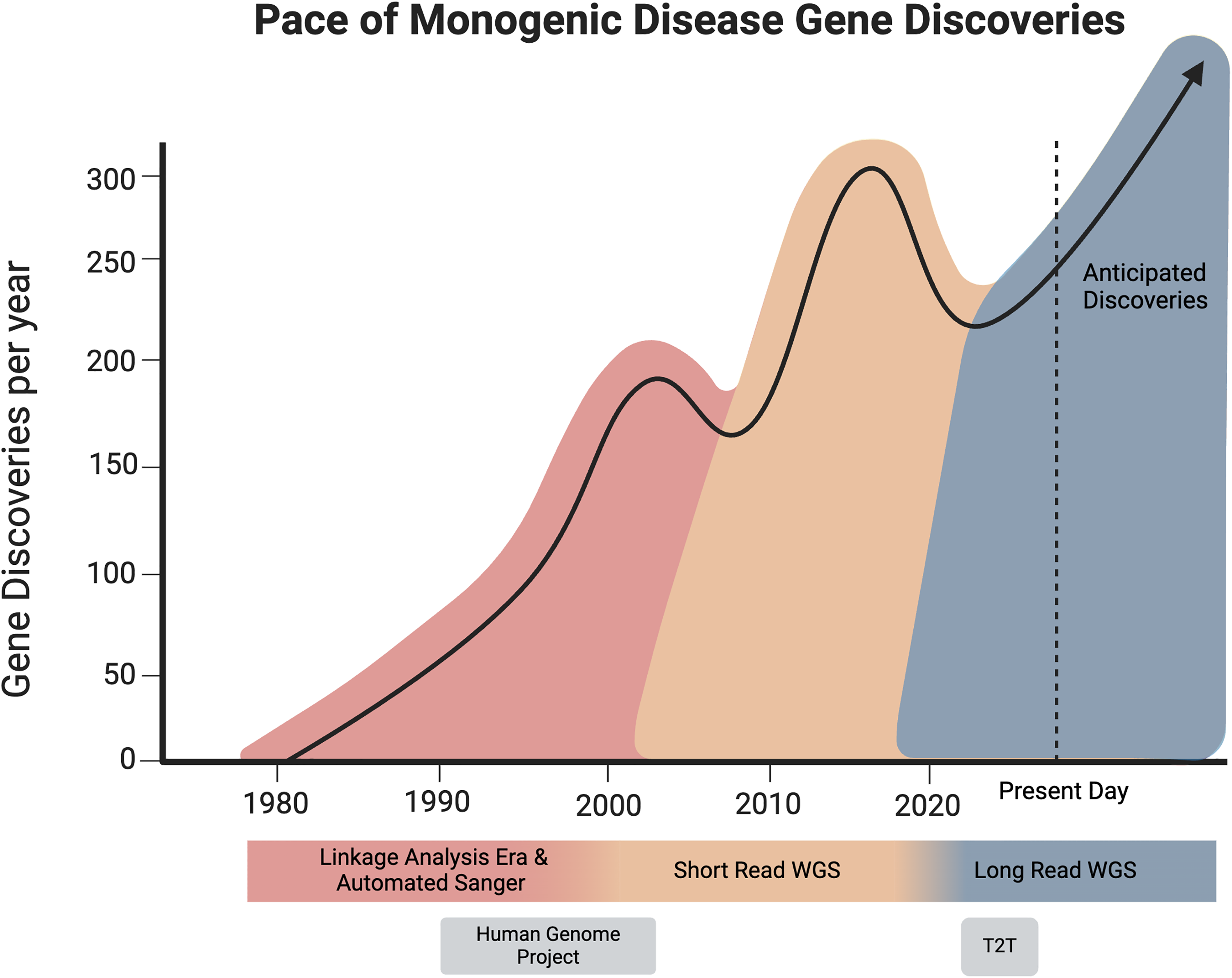
Diagram depicting approximate rate of gene discoveries and their coincidence with technological eras. Shortly following groundbreaking technological advancements, gene discovery spikes as research groups adapt to new technologies by developing software and optimize workflows and analysis methods.
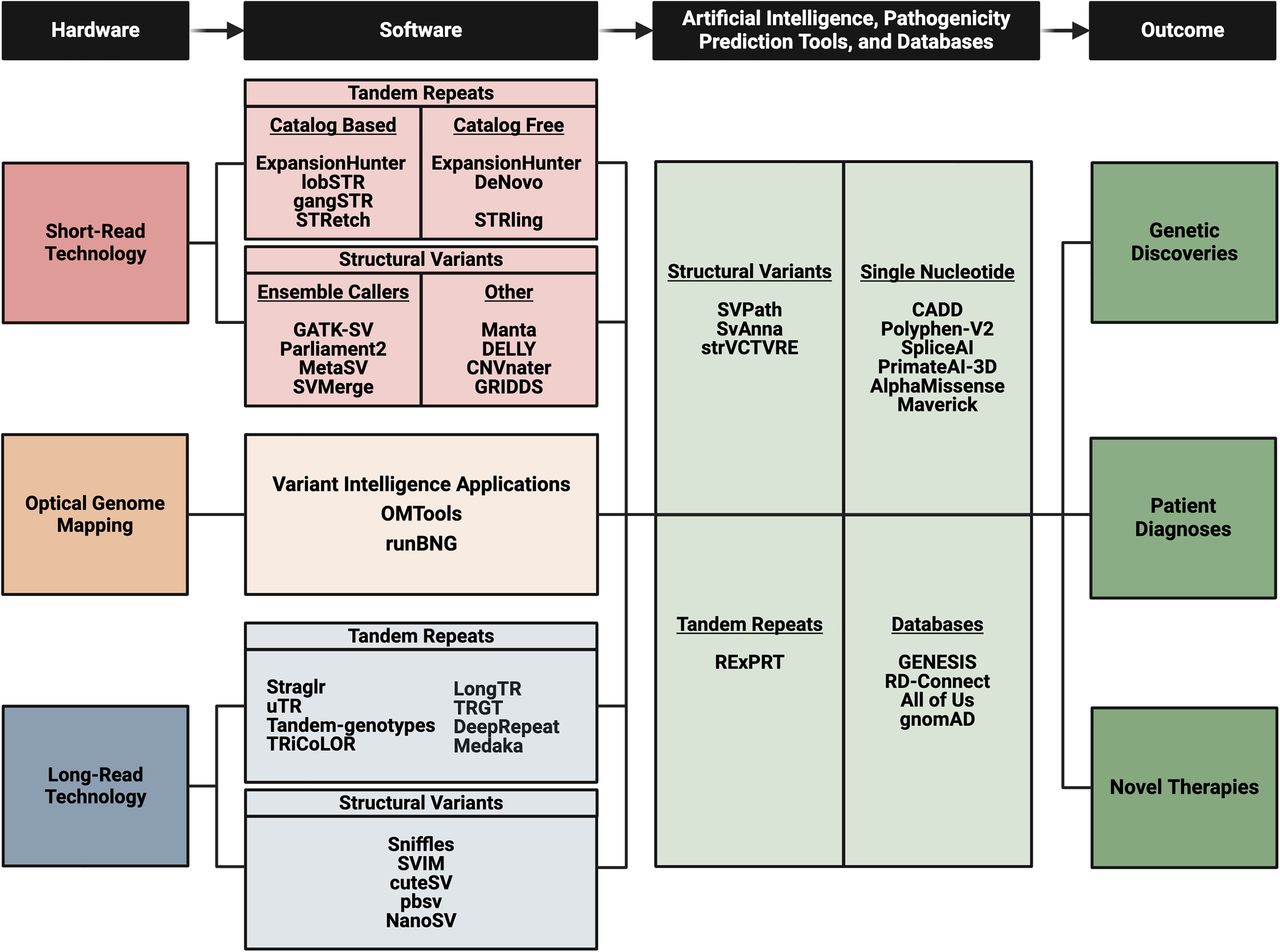
Diagram bulleting the various technologies and applicable software or tool options. As research labs begin implementing these practices for their rare neurogenetic disease cohorts there will be improved gene discoveries which will lead to patient diagnoses and future therapeutic endeavors.
The field of neurogenetics flourished with the advent of DNA sequencing techniques in the 1970s, notably Sanger sequencing, which enabled the reading of DNA sequences. The subsequent development of recombinant DNA technology in the 1980s facilitated the cloning and manipulation of specific genes, ushering in an era of isolated gene studies. Kerry Mullis’ revolutionary invention of polymerase chain reaction (PCR) in 1983 further accelerated DNA amplification, enabling the analysis of even minute DNA samples and aiding in genetic testing and diagnosis. 5
It was through these technologies that neurological phenotypes could be mapped to regions on chromosomes. Linkage analysis exploits the property that genes that are physically close to each other on chromosomes remain linked during meiosis. 6 Scientists would focus on genetic markers, such as microsatellites or single nucleotide polymorphisms, and track those that were consistently inherited along with the presence of the disease. In 1983, Huntington's disease became the first disorder to be molecularly mapped to a human chromosome using this technique. The localization of these genetic “linkage regions” allow for the eventual identification of the causal gene. 7 Ten years later, the Huntington disease Collaborative Research Group announced that a polyglutamine CAG trinucleotide expanded repeat was the cause of the disorder.8,9 Countless other disorders were similarly mapped and later isolated in this fashion, such as Charcot-Marie-Tooth disease Type 1A, Duchenne muscular dystrophy, and C9orf72 mediated Amyotrophic Lateral Sclerosis and Frontotemporal Dementia.6,10–14
Research involved in identifying genes associated with rare and common diseases is what inspired the Human Genome Project in 1990 – whose goal was to decipher the entire sequence of the human genome. This project lasted over a decade and amounted to nearly $3 billion. Sequencing the human genome created an enormous demand for DNA sequencing machines that were faster, cheaper, and provided higher accuracy. A breakthrough concept was the shotgun sequencing approach, whereby genomes would be assembled in computers out of millions of short sequencing fragments. 15 This led to the development of so called next-generation sequencing platforms that scaled the output of bases sequenced per ‘run’ by several orders of magnitude. 16 In combination with ingenious targeted capture methods, Exome sequencing was firmly established in 2009. 17 Further improvements led to routine Genome sequencing within a decade. Accordingly, the cost of sequencing a single human genome has dropped from >$1 M to <$500 in about 15 years. This has fueled gene discovery in the realm of neurological disorders with thousands of monogenic disease genes described. In 2024, there are over 80 hereditary spastic paraplegia, approximately 50 spinocerebellar ataxia, and more than 100 CMT associated disease genes.18–23 For complex neurogenetic disorders not solely caused by a single-gene mutation, genome-wide genotyping approaches extensively explored alleles that are associated with disease risk and or severity. Similar to linkage studies, non-random recombination patterns, or haploblocks, allowed single nucleotide polymorphisms (SNPs) to act as markers for nearby mutations that confer biological effects. With a large enough sample size, these association studies could identify SNPs that are statistically enriched in cases vs controls for example. These efforts have produced important insights into disease causation and also identified genetic modifiers of disease.24–27 However, the clinical application of such association studies is still being explored, with the introduction of ‘polygenic risk scores’ being the current peak of translational efforts. 28 Surprisingly to many observers, it wasn’t until 2022 when the Telomere-to-Telomere CHM13 (T2T-CHM13) reference genome was completed, which maps the entire genome for the very first time, including previously unresolved and complex regions. 29 The T2T reference and its multi-ancestral graph genome derivatives are the new gold standard enabling significant breakthroughs in diagnostics and treatment for neurological disorders. Importantly, many of the achievements built on multiple generations of statistical and bioinformatics software packages. In many ways, neurogenetics has become a data science with heavy reliance on computational methodologies (Figure 1).
Challenges for the next phase
Despite the success of NGS in discovering rare pathogenic variants and diagnoses, the current diagnostic gap is significant. For example, there are over 100 known CMT genes, yet over 50% of all patients do not have a genetic diagnosis today.30,31 This pattern is similar in most rare diseases.32–34 One reason may be technical limitations of short-read technologies. Current Exome and Genome technologies limit read lengths to 150 base-pairs on average and thus have difficulties detecting large structural variants including tandem repeat expansions, deletions, insertions, duplications, gene fusions, translocations, and other complex rearrangements.35–38 Another limitation of current neurogenetic and rare disease studies involve the difficulties in resolving variants of unknown clinical significance (VUS).39,40 Only a small percentage of variants are classified as either pathogenic or benign. Classifying tens of thousands of VUS remains an important ongoing challenge in human genetics. 41 Studies that aim to identify phenotype modulating genes require statistical approaches, filtering strategies, plus large patient and control cohorts. 42 This contradicts the nature of rare neurological disorders. In the following sections, we will discuss current technologies and approaches that may address these limitations and aid a new wave of gene discoveries and neurogenetic understanding.
Contemporary genome technologies: hardware
Short-read next generation sequencing, also known as 2nd generation sequencing, has dominated rare-disease research for the past decade. This is in large part due to base-calling accuracy, high throughput of these technologies, and dramatically lower costs. 43 Consequently, there exist a large array of computational tools and algorithms dedicated to short-read data analysis. Short-read sequencing is effective at studying single nucleotide and indel variants in most regions of the genome and has contributed significantly to finding the causal gene for thousands of Mendelian disorders.
The most common technology associated with short-read sequencing is Illumina's Sequencing by Synthesis (SBS) technology. SBS can produce libraries of paired and single end reads at uniform coverage. The Illumina instrument provides post-analytical quality evaluation and metrics of the data, allowing for the removal of low-quality reads. The importance and contribution of short-read NGS to neurogenetic research cannot be overstated. Many of the other major short-read sequencing providers use technologies derived from SBS, such as those provided by 10X Genomics and Element Biosciences, who mainly optimized the sequencing flow cell design rather than the underlying sequencing methodology. The main alternative for short-read sequencing is Sequencing by Binding (SBB) offered by Pacific Biosciences, which grants even greater read quality at the cost of reduced throughput per dollar.
Long-Read Sequencing, also known as 3rd generation sequencing, may address some of the current limitations of short-read sequencing.44–46 In contrast to the 150 bp read lengths that most short-read technologies employ, long-read technologies produce DNA reads over 10 kb in size. The two most common long-read technologies used for scientific research are Oxford Nanopore Technology (ONT) sequencing and PacBio single-molecule real-time (SMRT) sequencing. While previously criticized for low single nucleotide accuracy, modern PacBio and ONT machines reach >99% consensus accuracy. Long-read sequencing technology can be used to phase SNPs into haplotypes, improve the de novo assembly of genomes, and improve our understanding of genetic variation, mutation, and evolutionary processes. 45 The completion of the T2T genome was largely powered by the inclusion of these technologies. 47
Long-read sequencing has already been instrumental in discovering new disease-associated repeat expansions.48–50 Tandem repeats (TRs) are adjacent, repeating, nucleotide sequence motifs in our DNA. TRs are highly polymorphic and scattered throughout the human genome. There is evidence that TRs played an important role in the evolution of the human brain.51,52 The mutation rate of TRs is magnitudes higher than the standard genome mutation rate. 53 Due to this instability, TRs have the potential to expand in length. When specific TRs reach a size threshold that varies by disease, there can be pathogenic consequences. This phenomenon, known as repeat expansion, is associated with over 60 disorders that primarily affect the nervous system.54,55 Over half of these repeat expansion disorders have been discovered in the last decade as technology has improved. One recent example is the RFC1 repeat locus where biallelic expansions can lead to cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS). Wildtype AAAAG repeat motifs vary remarkably in size in the healthy population. However, for patients with CANVAS, AAGGG, ACAGG, AGGGC, and potentially other biallelic repeat expansions or compound heterozygous mutations cause this disorder.56–61 Adding to the complexity, certain motifs can demonstrate length- or configuration-dependent pathogenicity, as seen with AAAGG, which becomes deleterious only when exceeding 500 units. 57 Ultimately, the precise identification of pathogenic RFC1 repeat alleles, including their motifs and sizes, requires the resolution offered by long-read sequencing.
There is still a considerable gap in genome-wide knowledge regarding the variation in TR loci, repeat length, motif variation, and composition at population level. Resources such as TR-gnomAD and WebSTR provide TR length frequencies.62,63 However, they are limited by short-read technology. For instance, using short reads data and tools, expanded loci are likely underestimated in length and do not contain information about alternate motifs or repeat interruptions: crucial information when comparing to rare neurological disease patient data. The more recent TR discoveries illustrate that in many pathogenic loci, the range of healthy variation exceeds the length of short reads. Consequently, genotyping with short-read technology might not effectively differentiate lengths between patients and controls. We hypothesize that future TR discoveries will predominantly involve loci that have pathogenic length thresholds exceeding the resolution of short reads, especially in non-coding regions. Furthermore, while the flanking sequences of TRs can be aligned with short reads, the technology cannot characterize the composition of DNA enclosed within large TRs. For example, nested insertions or motif changes within large tandem repeats have been implicated in disease,64–66 yet are undetectable in short read data. It appears evident that discoveries such as RFC1 AAGGG repeat expansions causing CANVAS would have been more rapidly discovered with long-reads. This logic should apply to future tandem repeat discoveries. Long-read sequencing is ideally suited for characterizing when there is large heterogeneity in motif composition and length.
Long-read sequencing also can improve characterization of other forms of large structural variants (SVs) such as insertions, deletions, duplications, translocations, and inversions. SVs are genomic alterations of at least 50 bp in size and are the largest source of genomic variation in terms of percent DNA. Recent studies have highlighted their importance regarding health and disease or gene expression.67–69 It is estimated copy-number variants (deletions and duplications) are responsible for ∼15% of neurodevelopmental disorder cases.70–72 Short-read sequencing data often has difficulty detecting SVs larger than read length, especially for insertions. 73 For example, the 1.5MB duplication containing PMP22 associated with CMT1A is not reliably found by the most popular short-read structural variant calling tools. The exact breakpoint for SVs is also critical to interpreting pathogenicity; accurate resolution of breakpoints remains a challenge for many structural variant callers. 36 Long-reads will contain more information surrounding breakpoints, and will improve precision and recall, particularly for large repetitive regions of the genome. Some studies have shown long-read data can accurately report the breakpoint within 100 bp of the true breakpoint. 74 Further, reduction of false positive calls using long-read data will enhance discovery efforts and has already shown success in identifying complex SVs in mendelian disorders. 46 For rare neurological disorders in particular, SVs may be a source of missing heritability that has previously been understudied and unresolved.
Over 50% of the human genome is made up of repetitive sequences.75,76 Traditionally, highly repetitive sequences have been difficult to assemble with short-read technology. One example of a repetitive sequence we highlight is unprocessed pseudogenes. Unprocessed pseudogenes are the result of gene duplications and unequal crossing over. The second copy of the gene develops mutations over time that cause it to become non-functional and thus a “pseudogene” of the first copy of the gene. This form of pseudogene maintains the exon-intron structure, and thus have high homology to the parental gene. The most common recessive form of CMT2 is caused by mutation in the sorbitol dehydrogenase (SORD) gene.77,78 The most common CMT2 mutation in SORD is a frameshift (c.757delG) mutation that is fixed in the highly homologous proximally located SORD2P pseudogene, which introduced methodological complications during discovery. While this can prove a problem for PCR primer development, it may also interfere with sequence-read alignment and detection of structural changes in the SORD – SORD2P region. Alignment algorithms for short-read data will evenly distribute identical reads in a random manner, however when you have longer DNA reads such as with long-read data, there will be less identical reads, allowing for more accurate alignment. Long-read sequencing may improve detection of rare variation in these highly homologous loci, such as SORD, which will lead to enhanced diagnosis rates and future discovery.
In recent years, optical genome mapping (OGM) has emerged as a popular technology. 79 Bionano Genomics is a provider of OGM, marketing its Saphyr System for structural variant detection. The process begins with the extraction of high-quality, high-molecular weight DNA, followed by genome-wide fluorescent labeling of a 6 bp motif that occurs roughly 15 times per 100 kb. These tagged DNA molecules are then imaged in nanochannels revealing the unique patterns of fluorescent signals along the extended DNA molecules. Bioinformatics algorithms analyze these patterns to detect differences between samples, that may indicate structural changes of the genomic architecture. 80 OGM produces large DNA fragments over 200 kb in size, roughly 20x larger than that of many long-read technologies. Studies have highlighted the ability of these optical maps to identify chromosomal aberrations with 100% concordance. 80 This technology may be ideally suited for identifying more complex SVs that have multiple breakpoints and such as a deletion-inversion-duplication. OGM will assist researchers in identifying large DNA aberrations that are currently unidentifiable with short- and even long-read sequencing technology.
Contemporary genome technologies: software
Neurogenetic research has greatly benefited from the diverse array of software tools available to scientists and researchers. These tools empower investigators to efficiently analyze complex genetic data, uncover disease-causing variants, and gain valuable insights into the genetic basis of neurological disorders. Here we discuss some of the more popular or recent tools that have been developed and will continue to aid in future neurogenetic discoveries (Figure 2).
Accurate genotyping of tandem repeat size and structure is important because both pathogenicity and severity are typically length and motif dependent variables.81–83 Many tools have been created for this purpose. When using short-read sequencing data, ExpansionHunter, 84 lobSTR, 85 GangSTR, 86 and HipSTR 87 are tools for estimating allele size of short tandem repeats. Additionally, software such as STRipy have been designed to streamline analyses with user-friendly graphical interfaces for TR characterization and visualization. 88 While most short-read data tandem repeat calling tools can report alleles longer than read-length, repeats that are hundreds to thousands of base-pairs long are typically underestimated in size. These tools are also limited to pre-specified catalogs of repeat loci, typically derived from the motif and structure of the reference genome. Thus, these tools would be limited in their scope and ability to resolve unconventional alleles. ExpansionHunterDeNovo 89 and STRling 90 use a non-catalog based approach to determine repeats longer than read length. This provides the opportunity to identify non-reference motifs, non-canonical allele conformations, and novel loci. Still, exact repeat sizes and sequence composition remain uncertain. Long-read data is more reliable for genotyping the size of large repeats. New software tailored for long-read data can give accurate estimates of expanded repeats to the sequence resolution to allow for repeat interruption and allelic segmentation analysis. Tandem Repeat Genotyping Tool (TRGT) 91 is a new software designed for PacBio long-read genomes. TRGT also profiles mosaicisms, CpG methylation, and has a companion tool for visualization of reads overlapping the repeat.91,92 Straglr is a tandem repeat genotyping software that shows high accuracy with both simulated and patient Nanopore long-read data and can detect repeats at targeted (cataloged) loci and in non-targeted fashion. 93 DeepRepeat is a tandem repeat detection algorithm that uses nanopore ionic signals as input and applies a deep convolutional neural network to detect repeats through image classification. 94 Other popular tools for tandem repeat quantification in long-reads include Medaka (https://github.com/nanoporetech/medaka), LongTR, 95 uTR, 96 tandem-genotypes, 97 and TRiCoLOR. 98 Tandem repeat expansions are largely understudied due to technical limitations and may be the mechanism behind many unexplained rare neurological diseases. These software tools will aid in improving our understanding of tandem repeat variation in the human genome and identify tandem repeats potentially relevant to health and disease.
The importance of structural variation (SV) in neurological phenotypes is increasingly recognized. Algorithms for identifying SVs in short-read data involve looking for changes in read depth, identifying discordantly aligned paired-end reads or split reads, creating a de novo sequence assembly, or by applying a combination of these strategies. 35 However, identifying large and complex SVs remain a challenge with short-read data, and are prone to low sensitivity and high rates of false positive calls. Some popular software for identifying SVs in short-read data include Manta, 99 DELLY, 100 CNVnator, 101 and GRIDDS.102,103 Benchmarking of multiple SV calling software suggests that using an ensemble of software will help overcome individual limits and provide better overall detection. 35 Software such as GATK-SV, 104 Parliament2, 105 MetaSV, 106 and SVMerge 107 combine multiple software and unify results. In contrast, long-read data that spans 10 kb or more offer superior alignment to repetitive regions of the genome and have higher likelihood to span SV breakpoints with high confidence alignments. Specialized software tailored for detecting SVs in long-read sequencing data include Sniffles, 108 SVIM, 109 cuteSV, 110 pbsv developed by PacBio, NAHRwhals, 111 and NanoSV. 112 HiFiASM is a tool that uses long reads and performs haplotype-resolved de novo contig assembly. 113 These assemblies can then be compared to the reference to derive small and large variations using tools such as PAV or paftools. 114 Accurately being able to detect SVs with breakpoint resolution will improve interpretation of their molecular effects, and aid in future neurogenetic research studies.
Large structural re-arrangements such as translocations, inversions, and copy-number variants can transform the architecture of a genome. They can change the localization of genes, disrupt reading frames by splitting genes, or affect gene dosage which can have pathological consequences. Due to its ability to analyze DNA molecules exceeding 200 kb in length, optical genome mapping is superior to both short-read and long-read technologies in detecting these large genomic changes with low false positive rates. 79 Some notable tools dedicated to processing, analyzing and visualizing optical genome map data are OMTools, 115 and runBNG. 116 Bionano Genomics offers their own Variant Intelligence Applications (VIA) software that centralizes OGM workflow analyses as well as can merge combine NGS data or microarray data into one complete analysis. As technology such as OGM continues to advance, many discoveries in neurogenetics will be made.
Machine learning and pathogenicity prediction tools
An individual genome will contain millions of variants. At population level, rare variants are by far the most common type of variation; in fact, any nucleotide change compatible with life is currently carried by an individual of the human race.39,41 Thus, geneticists have put significant effort in trying to decipher, which variants are biologically meaningful and relevant for disease. Multiple software and tools have been developed to assist geneticists in predicting deleterious variants. Recently, AI and machine learning systems have been introduced for variant prioritization and interpretation.
Popular tools such as Combined Annotation-Dependent Depletion 117 (CADD) and PolyPhen-2 118 (Polymorphism Phenotyping Version 2) assign a score to predict the impact of amino acid substitutions on protein function based on multiple forms of annotation and evidence. SpliceAI is a deep neural network that predicts non-coding genetic variants that can produce cryptic splicing and may explain 9–11% of rare genetic disorders. 119 PrimateAI-3D is a 3D-convolutional neural network that reasons over 3D protein structures using variation common in nonhuman primates to predict those that are likely pathogenic in humans. 120 The rationale of PrimateAI-3D is that variants that are commonly observed in non-human primates are likely benign because they are tolerated through natural selection. Similarly to PrimateAI-3D, AlphaMissense is a deep learning model that uses AlphaFold2 protein structure predictions to infer pathogenicity of variants. 121 MAVERICK is an ensemble of transformer-based neural networks that can classify multiple forms of single nucleotide variants (SNVs) and indels to help infer pathogenicity. 122 MAVERICK is specifically designed for Mendelian rare diseases and can assess pathogenicity in both autosomal dominant and recessive contexts.
Tools like SVPath, 88 SvAnna, 89 and StrVCTVRE 90 play a crucial role in assessing the pathogenicity of structural variants, which have the potential to elucidate a significant portion of rare disease cases. As long-read sequencing technologies continue to enhance our ability to detect and resolve these complex structural variants, tools of this nature are poised to become increasingly valuable in advancing our understanding of the genetic basis of neurogenetic disease. RExPRT is a machine learning tool that can annotate and prioritize tandem repeats using the genomic position and motif. 123 RExPRT uses an ensemble supervised machine learning approach with support vector machines and extreme gradient boosted decision trees, which assign a score that estimates the likelihood of pathogenicity for a tandem repeat locus. As new long-read sequencing can better elucidate the true length and motif compositions of tandem repeats, RExPRT will serve as a valuable tool for both annotation and prioritization.
Only recently it became apparent that machine learning and AI-driven tools offer unique capabilities for unraveling the significance of millions of genetic variants, ultimately aiding in prioritizing variants and solving rare neurogenetic disorders.
Genomic databases and data sharing
Phenotype-genotype enrichment studies of rare variants are essential for genetic research of rare Mendelian neurogenetic disorders. However, some variants in disease genes will be so rare that they are private in any given cohort. Therefore, for neurogenetic studies involving rare disease, data aggregation is essential to increase statistical power in finding pathogenic variants and reaching a diagnosis. Here we highlight resources dedicated to this cause.
The Genome Aggregation Database (gnomAD) is a resource that aggregates Exome and Genome sequences from unrelated individuals and diverse ancestries.41,68,124 gnomAD recently released version 4.0.0 containing genomic data from over 800,000 individuals. This version contains roughly 138,000 individuals of non-European genetic ancestry. This diverse dataset is essential for researchers, allowing a more accurate assessment of variant frequencies and aiding in the identification of rare or population-specific variants associated with genetic disorders. One limitation of the current gnomAD dataset is the exclusive use of short-read sequencing technology, leaving uncertainty in structural variants, multinucleotide polymorphisms (MNPs), and SNPs within hard-to-sequence regions. A comparable data structure is provided by the Regeneron Genetics Center (RGC), which has produced a resource of nearly one million Exomes from diverse ancestral populations. This dataset is accessible through the RGC Million Exome Variant Browser and includes 20 million coding variants, with annotations highlighting those that may impact splicing or gene function. 125
The GENESIS platform (GENESIS) contains Exome and Genome data from unresolved rare disease patients with a variety of neurological phenotypes, including Charcot-Marie-Tooth disease, hereditary ataxia, spastic paraplegia, amyotrophic lateral sclerosis, hereditary dementia, etc. as well as unaffected family members. 126 As of April 2024, there are ∼ 20,000 datasets in GENESIS from over 20 countries with diverse ancestry. GENESIS allows for fast genomic analysis with advanced filtering methods and multiple tools and resources such as Maverick, CADD, gnomAD allele frequencies and constraint, as well as gene conservation. Deidentified data on GENESIS is kept secure, only available to academic users, and allows for genetic matchmaking. The GENESIS platform has aided in the discovery of over 100 novel Mendelian disease genes since 2011.127,128
Rare-Disease Connect (RD-Connect) is a European infrastructure project for rare disease research. RD-Connect, similar to GENESIS, offers a data platform with a sizable number of bioinformatic tools assisting researchers with linking genetic variation to clinical phenotypes.129–131 RD-Connect has been a broadly used tool by the neurogenetic community. The Solve-RD consortium, utilizing the RD-Connect platform, has been pivotal in resolving 29% of reanalyzed cases and uncovering novel pathogenic mobile element insertions and structural variants in previously undiagnosed patients.132–134
The UK Biobank represents another key initiative in data aggregation. Recently, this initiative produced a dataset of 500,000 whole genomes with an average coverage of 32.5x, significantly expanding the scope of earlier exome-based databases. 135 Accessible through a secure, cloud-based platform, it fosters research collaborations across academia, industry, charities, and government sectors. As the largest genome sequencing effort to date involving individuals of non-European ancestry, the UK Biobank has already revealed novel genetic signals in Asian and African populations, further advancing our understanding of global genetic diversity.
The All of Us research program (AOURP) is an initiative to sequence at least one-million participants with diverse ancestral backgrounds residing in the USA and allow research access with the goal of advancing precision medicine and human health.136,137 As of April 2024, the All of Us data browser contains electronic health records and genomic sequencing data from ∼500,000 participants, including many rare neurological disease patients. The AOURP offers a cloud-based computing Researcher Workbench and cohort builder to allow bioinformaticians secure access to this large resource of data and analytical tools. 138 A pillar of the AOURP is the inclusion of ancestral backgrounds that are less commonly studied in science. Significantly, the AOU data browser provides a growing data resource using long-read PacBio and Nanopore technology, particularly from underrepresented backgrounds. This is crucial for establishing a normative control database of high quality structural variant calls. Data aggregation initiatives such as these will cluster patients with similar phenotypes, enhancing research efforts to discover the pathophysiology underlying neurogenetic disease.
Conclusions
In conclusion, the field of neurogenetics has witnessed a remarkable transformation driven by the rapid evolution of genomic technologies. These advancements, including long-read sequencing, optical genome mapping, and artificial intelligence, promise to help us better understand the connections between genes and neurological disorders. As we reflect on the history of genomic technologies in neurogenetics and the challenges faced in diagnosing rare neurogenetic disorders, it becomes clear that modern genomic solutions are poised to further shape our understanding of the human condition.
Long-read sequencing, with its ability to capture large structural variants and tandem repeats, stands as a potential game-changer in gene discovery and genetic diagnostic efforts. Optical genome mapping offers a holistic view of genomic architecture, especially valuable in identifying complex structural variations. Software tools equipped with AI and machine learning capabilities enhance our capacity to prioritize variants and unlock the mysteries of neurogenetic disorders.
Moreover, data aggregation initiatives like GENESIS, RD-Connect, UK biobank, and the All of Us research program empower researchers to gather diverse datasets, fostering collaborations that are vital for unraveling the genetic underpinnings of rare diseases. In this era of abundant genomic data, these tools and resources provide a pathway towards addressing the challenges posed by millions of genetic variants and, ultimately, for advancing the diagnosis and treatment of rare neurogenetic disorders.
Footnotes
Acknowledgments
The authors are supported by grants from MDA, CMTA, CMTRF, and NINDS (5R01NS105755, 5R01NS072248, 5U54NS065712, 5U24NS120858 to SZ).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.