
Abstract
Background:
Inflammatory myopathy with abundant macrophage [IMAM] is marked by macrophage infiltration and muscle fibers damage, resembling dermatomyositis [DM] but with unique pathology. Its mechanism remains unclear. Our study focused on exploring the clinicopathological characteristics, underlying pathogenic mechanisms, and the challenges in diagnosing and managing IMAM.
Methods:
A systematic analysis of medical literature databases (Pub-med, Cochrane, Scopus, Google Scholar) was performed using the term “IMAM,” excluding studies on other inflammatory myopathies [IMs]. Selected studies were independently assessed with the Newcastle-Ottawa Scale, and quantitative data underwent inter-statistical analysis, descriptive and odds ratio, to identify relevant findings.
Results:
Eight studies, including 49 IMAM cases from 2003 to 2024, were analyzed. Five were case reports, and three were cross-sectional studies. IMAM showed no age or sex predilection. Common symptoms included proximal muscle weakness, pain, and fatigue, with atypical DM-like skin features in 65% of cases. Other association included hemophagocytosis, cutaneous panniculitis, and interstitial lung infiltration. Histologically, all cases showed myonecrosis infiltrated with CD68+ macrophages. Scattered CD3+ and CD4+ T-cells expressing IL-10 with no or rare CD8+ T-cells were identified. MAC deposition was limited to necrotic fibers, and perifascicular atrophy was absent in all cases. Anti-PL-7 and anti-U1 RNP antibodies were detected in 4% of cases. Elevated TNFα and IFN-γ levels, with low STAT1 and STAT6, were observed. Genetic analysis revealed MEFV polymorphisms in 7 cases and a TNFRSF1A mutation [C43R] in single case. Treatment involved steroids, with or without immunotherapy or chemotherapy, leading to remission and recovery in 43.7% of cases.
Conclusion:
IMAM is a distinct type of IMs that requires muscle biopsy for diagnosis as myositis antibody and cytokine tests are usually insensitive.
Introduction
In addition to the classical idiopathic inflammatory myopathies [IIMs], a new spectrum of inflammatory myopathy with predominant macrophage are explored in the literature including inflammatory myopathy with abundant macrophage [IMAM], 1 macrophagic myofasciitis [MMF] 2 and post-vaccine myositis [PVM]. 3 While PVM are commonly encountered as myositis due to vaccination effect, IMAM is considered a distinct entity in which its mechanism remains uncertain. 4 IMAM also differs from MMF, which presents with chronic arthromylagias without muscle weakness, in which the pathology is connected to aluminium hydroxide- containing vaccines. 5 The main remarkable features with MMF are macrophage collection, lymphocytic influx that showed strong CD8+T-cell expression and ultrastructural collection of intracytoplasmic aggregates of fine needle-shaped clusters. 6 Besides MMF or dermatomyositis [DM], difficult diagnostic problems were encountered in patients in whom muscle biopsy showed huge infiltrations of fascia by large macrophages, previously unreported in these settings.
IMAM was first described in French population by Bassez et al. in 2003 when they found predominant macrophages in 27 cases of atypical DM. 1 Sequence of cases were reported later identifying the disease patterns [Table 1]. IMAM is characterized by diffuse infiltration of macrophages in the skeletal muscle and fascia associated with muscle fibers damage. 1 The condition is expressed as DM-like disease, but distinct from DM pathology. 4 IMAM is clinically presented with mild-to moderate muscle weakness involving proximal extremities more than the trunk and associated with typical or atypical DM-like skin alterations, which frequently misdiagnosed as DM or overlap myositis syndrome 1 [Table 2]. Some patients typically have no distinct clinical features, or they present with mild muscle pain and weakness. Creatine phosphokinase [CKP] levels in IMAM can vary from normal to elevated, but generally do not exceed 5000 U/L. 6 Although the diagnostic sensitivity of magnetic resonance imaging [MRI] for IMAM has yet to be assessed, reported cases have shown thickening, hypertrophy, and edema in both the fascia and muscle tissues. 7 Electromyography [EMG] can reveal abnormal myogenic changes, including fibrillation in affected muscles. 7 Myositis-specific antibodies [MSA] typically found in DM, such as anti-Mi-2, anti-Ku, and anti-PM-Scl, are infrequently detected in IMAM [Table 2]. Nonetheless, testing for these antibodies is essential to minimize the differential diagnoses for this pathological condition.
Published studies of IMAM and the New-castle Ottawa scale for quality assessment of selected studies.
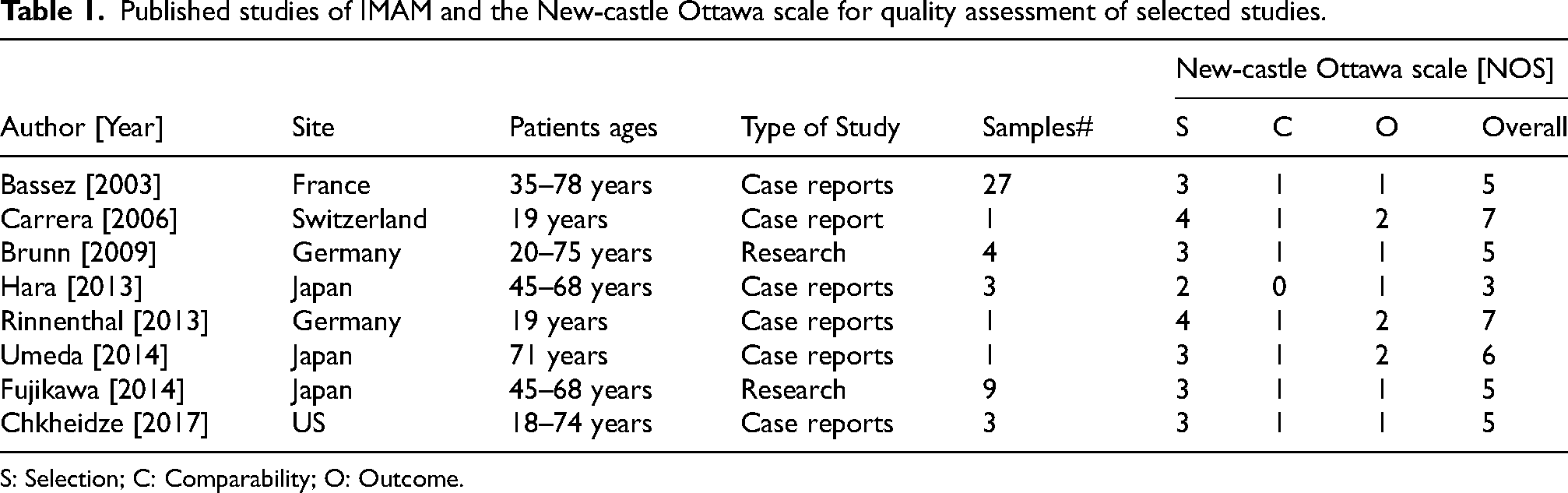
S: Selection; C: Comparability; O: Outcome.
Clinical, laboratory and pathological findings as well as other abnormal association and treatment approaches of IMAM.

The best method to confirm the diagnosis is muscle biopsy. IMAM is histologically characterized by the presence of myopathic features, myonecrosis, and chronic inflammation. 1 The leukocytic inflammatory infiltrates in IMAM are primarily located at the periphery of muscle fascicles and consist mainly of CD68+ macrophages [Table 2] [Figure 1]. These macrophages are not those surrounding necrotic fibers but are instead concentrated predominantly in the perimysial and epimysial regions, with a lesser presence in the endomysial connective tissue spaces. 8 IMAM macrophages are more dispersed, lack basophilic PAS-positive granules, stain positive with acid phosphatase, but are negative with Morin staining. 9 Additional histological features include predominant CD4+T-cells, rare CD8+T-cells, and membrane attack complex [MAC] deposition restricted to necrotic fibers. 4 The absence of perifascicular atrophy, intact endomysial capillaries, and MAC [C5b9] deposition only on necrotic fibers are distinguishing characteristics of IMAM compared to DM. 4
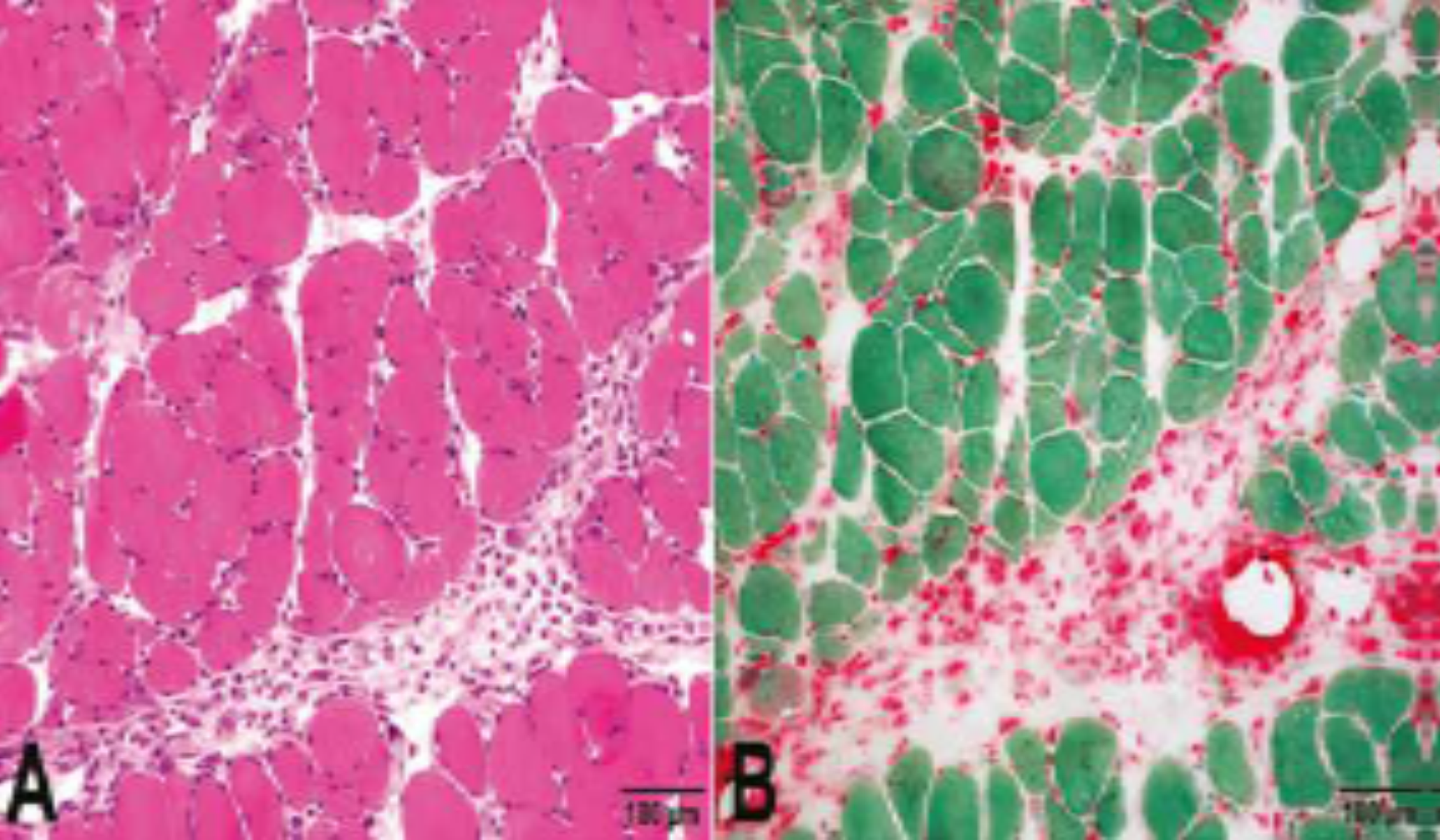
Hematoxylin and eosin (a) and CD68 (b) staining of a section from an IMAM case shows myopathic features with predominant macrophages in the perimysial spaces (x100 μm).
In this research, we systematically examined eight published studies on IMAM, providing a summary of the clinical, pathological, and prognostic findings, as well as an overview of potential pathogenic mechanisms associated with the disease. This review aims to bridge critical gaps in understanding IMAM by synthesizing current evidence, highlighting its clinical relevance, and emphasizing its importance for improving diagnostic accuracy, future management, and guiding future therapeutic strategies.
Methodology
Data search
A systematic search was conducted in literature databases including PubMed, Cochrane, Google Scholar, and Scopus using the search string ‘inflammatory myositis with abundant macrophages, “IMAM”, macrophagic myopathy’. Additional research and review articles were manually searched. No restrictions were applied regarding language or publication year. All studies on IMAM focusing on clinical presentation, pathogenesis, diagnosis, pathological findings, prognosis, and treatment were included, while studies centering on other IMs or types of myopathies with macrophage involvement, lacking any keywords from the search string, were excluded. The identification and selection process followed the PRISMA guidelines [Figure 2].
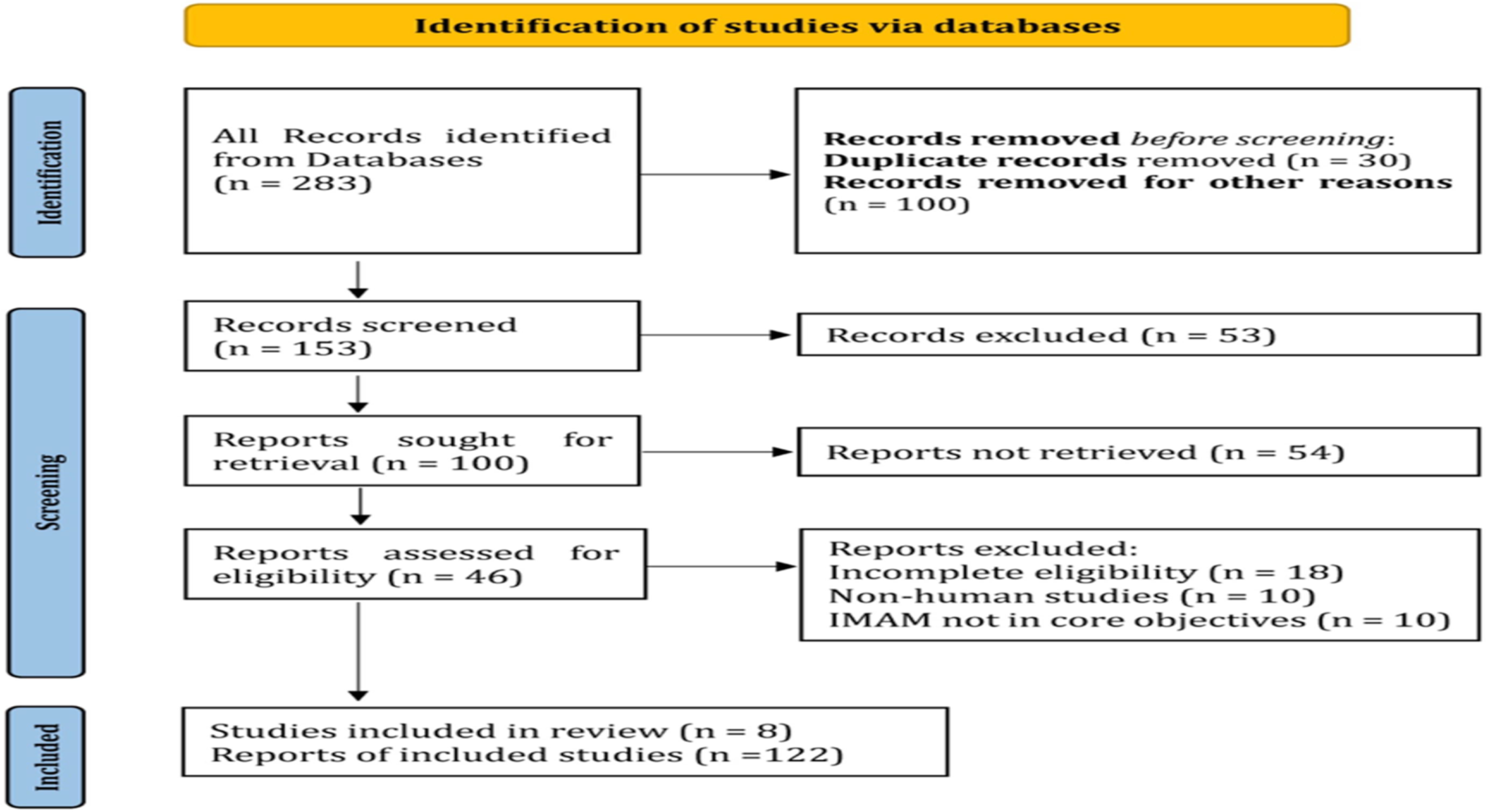
PRISMA flow diagram demonstrating study selection.
Data extraction and quality assessment
The extracted articles were organized into categories to reduce confusion: (1) clinical presentation and causes, (2) diagnosis and pathological findings, and (3) treatment and prognosis. Following a thorough review of the selected studies, core data were recorded, including author [year], site, study type, and number of IMAM cases [Table 1]. Data extraction was conducted by two authors [AB] and [MK]. Each included study was independently evaluated and the Risk of bias assessed using the Newcastle-Ottawa Scale [NOS], a tool for assessing the quality of non-randomized studies, particularly cohort and case-control studies in systematic reviews and meta-analyses. The NOS uses a star rating system to evaluate quality across three main aspects: [1] selection of study groups, [2] comparability of groups, and [3] outcome determination, with scores ranging from 0 to 9 stars [Table 1].
Statistical analysis
The strength of association of common pathological findings with IMAM were evaluated using descriptive values while Odds ratio with 95% confidence interval (CI) was used for some features. The analysis was performed by IBM SPSS V.27. Analysis was applied on the data that was quantitatively reported in the selected studies.
Results
Eight studies conducted between 2003 and 2024 reported a total of 49 cases of IMAM. Of these, five were case studies, and three were cross-sectional studies based on sample data [Table 1]. IMAM showed no specific age or sex predilection, with cases involving patients aged between 18 and 75 years.
Clinical presentation and causes
The predominant clinical presentations were muscle weakness and pain, primarily affecting the proximal extremities rather than the trunk1–5 [Table 2]. Typical or atypical DM-like skin alterations were identified in 65% of cases, often leading to misdiagnoses as DM or overlap myositis syndrome.1–5 Hemophagocytosis was observed in about 60% of IMAM cases, making it a common associated feature.1,4 Rare associations included cutaneous panniculitis and interstitial lung infiltration.7,10–12 Sweet's syndrome, characterized by fever, painful skull lesions, arthralgia, and, rarely, conjunctivitis, was reported in one patient. 9 Severe forms of IMAM were associated with macrophage activation syndromes [MAS]. 13 Myositis-specific antibodies [MSA] typically found in DM, such as anti-Mi-2, anti-Ku, and anti-PM-Scl, were generally absent in IMAM, although only two cases were reported [4%] with anti-PL-7 and anti-U1 RNP antibodies 7 [Table 2].
Diagnosis and pathological findings
The primary specimen for IMAM research was biopsied muscle tissue, and histological analysis showed myonecrosis and abundant CD68+ macrophages, with scattered CD3+ and CD4+ T-cells expressing IL-10. CD8+ T-cells and CD20+ B-cells were rare, appearing in only three cases, and CD68+ macrophages strongly expressed MRP14+ 4 [Table 2]. CD123-expressing plasmacytoid cells were also detected as a characteristic feature of IMAM. Single-fiber necrosis and microinfarcts were significantly more prevalent in IMAM compared to standard IMs. 1 MAC [C5b9] deposition was limited to necrotic fibers, with no perifascicular atrophy identified in any cases. 4 Ultrastructural analysis of capillary endothelial cells generally did not reveal tubuloreticular inclusions [TRI], except in one case.8,9 Non-specific ultrastructural findings included myelin figures and blood cell phagocytosis. 1
Elevated levels of TNF-α and IFN-γ, along with high expression of Signal Transducer and Activator of Transcription 1 (STAT1) and STAT6, are hallmark features in IMAM.1,4,8 These cytokines and transcription factors reflect the disease's macrophage-driven inflammatory response and immune dysregulation. TNF-α and IFN-γ are key pro-inflammatory mediators driving macrophage activation and muscle damage, while STAT1 supports a Th1 immune response, and STAT6 promotes M2 macrophage polarization. Together, these markers indicate a mixed inflammatory environment. Genetic analysis identified MEFV polymorphisms [G304R, R202R, E148Q, E148Q-L110P, and P369S-R408Q] in 7 patients, while single IMAM patient had a TNFRSF1A mutation. 6
Treatment and prognosis
Treatment primarily involved steroids, with or without immunotherapy or chemotherapy, resulting in remission in 43.7% of cases. IMAM is typically differentiated from other IMs, such as DM and MMF, at the histological level by key features including leukocytic infiltration, hemophagocytosis, CD8+ T-cells, and MAC [C5b9] deposition.1–3 There was a 3.76-fold higher chance of leukocytic infiltration in biopsied muscle tissues of IMAM compared to those of DM or MMF. Hemophagocytosis was observed in IMAM muscle samples at odds 26.6 times higher than in other IM muscle samples, while other IMs, including MMF, were found to have 1.5 times [1/0.67] more CD8+ T-cells than IMAM. The clinical features, workup and pathological findings, and other association are summarized in Table 2.
Risk of bias
The NOS was utilized to assess the quality of the included studies, focusing on selection, comparability, and outcome [Table 1]. Case reports generally scored higher in the selection and outcome domains but lacked comparability due to their observational nature. Cross-sectional studies provided more extensive data but scored lower on comparability due to methodological variability. Outcome scored as 1 is indicating minimal validation of outcomes, reliance on subjective or non-standardized measures, or inconsistent reporting. Lower scores (e.g., Hara, 2013) reflect deficiencies across domains, including unclear selection methods and no confounder adjustment, while higher scores (e.g., Rinnenthal, 2013) suggest robust methodologies. Most studies lacked rigorous outcome assessment, limiting reliability.
Overall, the limited sample size and inherent variability across studies contributed to a moderate risk of bias, emphasizing the need for cautious interpretation of results and further research with standardized protocols.
Discussion
The link between DM and IMAM is indicated in a patient diagnosed with DM for few months, and later, a repeated biopsy revealed focal IMAM damaging the muscle. 10 This phenomenon may reflect a superimposed acute-on-chronic form of DM. Brunn et al. found that three out of four IMAM cases were initially misdiagnosed as DM based on early muscle biopsies. 4 This misdiagnosis often occurs when patients present with DM skin features, but early biopsies show no inflammatory macrophages. Diagnosing IMAM begins with clinical evaluation of the patient presenting with proximal muscle weakness, myalgia, and occasional DM-like skin changes.1,4 Systemic symptoms, including fever and arthralgia, may also occur. Blood investigations reveal elevated inflammatory markers, creatine kinase (CK), and increased level of ferritin, alongside increased cytokines like TNF-α and IFN-γ, indicating inflammation and muscle damage. These findings provide initial clues, guiding clinicians toward muscle biopsy, the gold standard, to confirm IMAM and distinguish it from other inflammatory myopathies.1,4,6,8–14
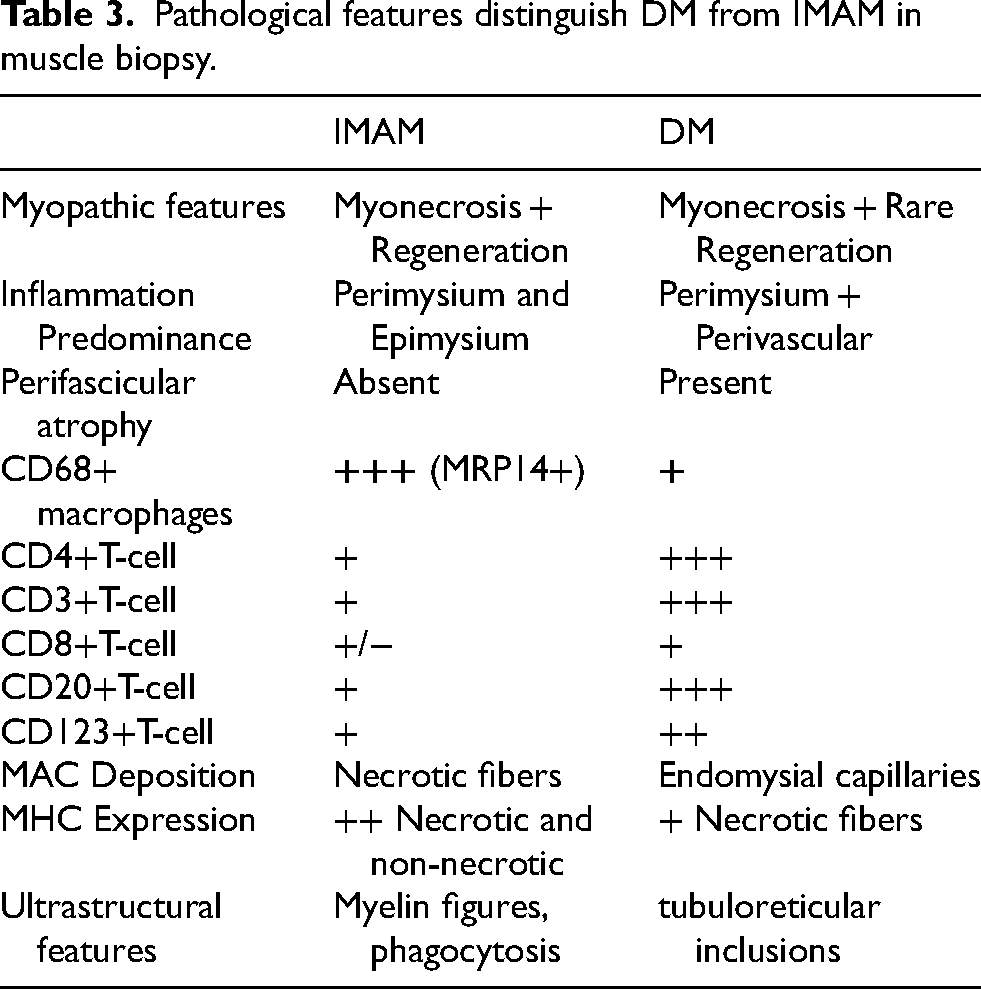
IMAM is generally characterized by muscle fiber damage and a chronic inflammatory leukocytic infiltrate, predominantly CD68+ macrophages at the periphery of muscle fascicles, with these macrophages expressing unique markers like MRP14+ and 25F9+.4,14 MRP14+, also known as S100A9, is expressed by neutrophils and certain macrophage subsets, playing a role in regulating inflammation and immune recruitment to affected tissues. 15 While macrophages in DM may resolve with treatment, macrophages in IMAM appear persistent, marked by 25F9+ inflammatory macrophages seen in the perimysium in later disease stages, contributing to chronic inflammatory responses and tissue remodeling.14,16 IMAM differs from DM, where CD4+ T cells are commonly present, but CD68+ macrophages and CD8+ T cells are minimal or absent, with rare occurrences of CD3+ and CD4+ T cells. 4 The absence of perifascicular atrophy, intact endomysial capillaries, and MAC [C5b9] deposition only on necrotic fibers help distinguish IMAM from DM. 4 Regarding MHC-Class I expression, DM exhibits diffuse upregulation on non-necrotic fibers, whereas IMAM shows focal, weaker expression on necrotic fibers. 9 Features distinguish IMAM from DM are summarised in Table 3. Hypoxia markers, such as HIF-1α and CD206, were highly expressed in IMAM-affected muscle fibers, indicating hypoxia-related pathology, 8 while the lack of MAC in muscle sarcoplasm suggests IMAM may not be microvasculopathic in origin. 17 IMAM shares features with cytophagic histiocytic panniculitis [CHP], an autoimmune disorder affecting multiple organs and potentially leading to hemophagocytosis and pancytopenia.18,19 Both IMAM and CHP, unlike macrophagic myofasciitis [MMF], may be associated with life-threatening hemophagocytic syndromes, warranting further investigation into overlapping blood and histological abnormalities to rule out CHP.
Pathological features distinguish DM from IMAM in muscle biopsy.
Possible pathogenesis and related machanisms
IMAM may involve CD4+ T cell clones that enhance the expression of cytokines such as IFN-γ and TNF-α.1,7,8 Elevated levels of TNF-α, IFN-γ, STAT1, and STAT6 in skeletal muscle and blood were observed in IMAM cases before treatment,7,8 with these biomarkers acting as critical regulators of inflammation and immune signaling. 20 TNF-α binds to muscle and immune cell receptors, activating NF-κB to induce pro-inflammatory cytokines, chemokines, and adhesion molecules, promoting muscle apoptosis and immune cell recruitment, thereby sustaining inflammation. 21 IFN-γ, produced by T cells and NK cells, activates macrophages, enhancing antigen presentation and upregulating MHC class I on muscle cells, making them CD8+ T cell targets and fostering Th1 immune responses that contribute to muscle damage. 22 IMAM's elevated TNF-α and IFN-γ levels promote CD68+ macrophage recruitment and activation, and reduces CD8+ T-cell activity, emphasizing a macrophage-dominant immune response unique to IMAM compared to other myopathies. IMAM's immune response differs from other myopathies as these pro-inflammatory cytokines drive tissue damage and inflammation. Unlike DM, IMAM rarely involves autoantibodies, making serological tests less useful, and relies heavily on muscle biopsy for diagnosis. This distinct pathology explains IMAM's limited response to steroid and immunotherapies. Therefore, targeted therapies addressing macrophage activation, such as TNF-α inhibitors, are needed to improve treatment outcomes, highlighting its unique diagnostic and therapeutic challenges.
Myositis-specific antibodies [MSA] typically found in DM, such as anti-Mi-2, anti-Ku, and anti-PM-Scl, are generally absent in IMAM, though Hara et al. reported two cases with anti-PL-7 and anti-U1 RNP antibodies. 7 The association of these antibodies with IMAM remains unclear. Anti-PL-7, an autoantibody targeting threonyl-tRNA synthetase, is usually associated with antisynthetase syndrome [ASSD], interstitial lung diseases [ILDs], and has been investigated in DM.23,24 Anti-U1 RNP is a marker for mixed connective tissue diseases and has been observed in DM but was not previously reported in IMAM, except in rare cases.7,25 Thickened connective tissue spaces with macrophages and mast cells infiltration have been observed in these conditions.
In autoinflammatory syndromes linked to innate immunity genetic variants, macrophages play a pivotal role by releasing diverse cytokines. Familial Mediterranean fever [FMF], febrile myalgia syndrome [FMS], and TNF receptor-associated periodic syndrome [TRAPS] are known to cause severe myalgia.26,27 FMF is caused by MEFV gene mutations, while TRAPS results from TNFRSF1A gene mutations.28,29 Fujikawa et al. explored these connections, identifying MEFV polymorphisms [G304R, R202R, E148Q, E148Q-L110P, and P369S-R408Q] in seven IMAM patients and a TNFRSF1A mutation in one patient, suggesting that MEFV polymorphisms and TNFRSF1A mutations may serve as susceptibility and modifier genes in IMAM, warranting further investigation in larger cohorts. 6
Treatment options and prognosis
Treatment protocols used in IMAM commonly involve prednisone [1 mg/kg per day] and azathioprine [1.5–3 mg/kg per day].6,10 In cases of relapse, additional investigations such as serum ferritin, bone marrow analysis, and T-cell clonality are recommended to exclude hemophagocytic syndromes and CHP.
7
Severe IMAM forms have been linked to MAS, likely caused by uncontrolled T-cell and macrophage proliferation, either as a primary condition or secondary to malignancy. Resistance may stem from persistent macrophage-driven inflammation, necessitating adjunct therapies like plasmapheresis or immunosuppressants. Intravenous immunoglobulin (IVIG) has been reported as a potential adjunct therapy in IMAM, particularly in cases refractory to steroid treatment or those associated with MAS. IVIG may modulate immune responses by reducing cytokine levels like TNF-α and IFN-γ, which drive macrophage activation (
High ferritin levels and muscle fiber degradation products are alert signs in IMAM cases. Among nine cases reported by Fujikawa et al., 6 cases showed remission, while 3 cases improved significantly with steroid, immunotherapy, or chemotherapy. 6 In such cases, methylprednisolone, plasmapheresis, leukocytapheresis, and polymyxin B-immobilized fiber hemoperfusion may be recommended. Immunosuppressants such as tacrolimus or cyclosporine can be administered to stimulate bone marrow response, which can be monitored by TNF-α and sTNFR1 levels in the blood. 7 Where these markers are unavailable, regular blood tests and patient observation are generally sufficient. Remission or improvement was estimated to be in 43.7% among all cases.
Limitation
This study's limitations include a small sample size, reliance on case reports, and variability in reporting methods. Statistical strength is reduced by missing measurements. Larger, standardized studies are needed to validate and generalize findings
Conclusion
IMAM is a rare, distinct subtype of inflammatory myopathy that should be carefully differentiated from DM, particularly in patients with DM-like skin features. Muscle biopsy remains the gold standard for diagnosis, typically revealing CD68+ macrophage infiltration, CD4+ T-cells, and a lack of CD8+ T-cells, CD20+ B-cells, perifascicular atrophy, with MAC deposition restricted to necrotic fibers. Despite limited published studies on IMAM, further case studies and research should be reported and combined with comprehensive pathological, immunological, and genetic investigations.
Footnotes
Acknowledgment
None.
Author contribution(s)
All authors have critically edited, reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
N/a.
Informed consent
Informed consent is not required in this study.
Availability of data and material
The datasets generated during and/or analysed during the current study are available from the corresponding author [MK] on reasonable request.