
Abstract
X-linked myopathy with excessive autophagy (XMEA) is a rare neuromuscular disorder caused by mutations in the VMA21 gene, encoding a chaperone protein present in the endoplasmic reticulum (ER). In yeast and human, VMA21 has been shown to chaperone the assembly of the vacuolar (v)-ATPase proton pump required for the acidification of lysosomes and other organelles. In line with this, VMA21 deficiency in XMEA impairs autophagic degradation steps, which would be key in XMEA pathogenesis. Recent years have witnessed a surge of interest in VMA21, with the identification of novel mutations causing a congenital disorder of glycosylation (CDG) with liver affection, and its potent implication in cancer predisposition. With this, VMA21 deficiency has been further linked to defective glycosylation, lipid metabolism dysregulation and ER stress. Moreover, the identification of two VMA21 isoforms, namely VMA21-101 and VMA21-120, has opened novel avenues regarding the pathomechanisms leading to XMEA and VMA21-CDG. In this review, we discuss recent advances on the clinical spectrum associated with VMA21 deficiency and on the pathophysiological roles of VMA21.
Introduction
During the past decades, genetic studies in humans have increasingly linked insufficient lysosomal acidification to pathological conditions, such as neurodegenerative disorders, cancer, inflammatory diseases and myopathies.1–4 Impaired lysosomal acidification results in the accumulation of toxic substrates by disrupting autophagy, a catabolic process engulfing and ultimately degrading cytoplasmic components and organelles. 5 Lysosome acidification, sustaining the hydrolytic activity of luminal enzymes, involves the vacuolar ATPase (v-ATPase) proton pomp at the lysosomal membrane. Assembly of the two large domains forming the v-ATPase occurs sequentially in the endoplasmic reticulum (ER) and Golgi, and is controlled by chaperone proteins, such as TMEM199, CCDC115 and VMA21. Mutations in genes encoding sub-units or chaperones of the v-ATPase cause various diseases, associated with lysosomal acidification impairment. 6 In particular, mutations in the VMA21 gene has been identified in 2013 as the genetic cause of a rare neuromuscular disorder, called X-linked Myopathy with Excessive Autophagy (XMEA), 7 and several years later of a congenital disorder of glycosylation (CDG) affecting the liver. 8 More recently, VMA21 variants were associated with follicular lymphoma (FL), highlighting for the first time the role of VMA21 in cancer development. 9 Impaired autophagy, consistent with a mis-assembly of the v-ATPase, has been incriminated as one of the key pathomechanisms of VMA21-related diseases. However, autophagy defect cannot solely explain the heterogeneous clinical spectrum and the tissue specificity observed in these diseases. The recent discovery of distinct VMA21 isoforms also raises questions on their respective pathophysiological roles in the disorders. In this review, we dissect the clinical features of XMEA, CDG and FL, and we compare the pathomechanisms that may contribute to the pathogenesis of these diseases. Moreover, we provide a complete list of the mutations reported in the three conditions, and discuss their functional consequences and the associated clinical diversity.
VMA21-related disorders: from genetic basis to clinical diversity
XMEA: a rare autophagic vacuolar myopathies
Autophagic vacuolar myopathies (AVMs)
AVMs constitute a group of neuromuscular diseases characterized by autophagic vacuoles in skeletal muscle biopsies from patients. These vacuoles accumulate in muscle fibres and stain positive for autophagic and lysosomal protein markers. They commonly originate from a dysregulation of the lysosome-autophagy process, although the pathomechanisms differ from one disease of the family to another. 10
A first group of AVMs is caused by primary lysosomal dysfunction. The two most prevalent AVMs of this group are Pompe disease (or glycogen storage disease II, GSDII) and Danon disease (or glycogen storage disease IIb, GSDIIb). Pompe disease is caused by mutations in the GAA gene, which encodes the lysosomal enzyme acid alpha-glucosidase (GAA), essential for the degradation of glycogen to glucose. In Pompe disease, GAA deficiency results in the accumulation of glycogen in lysosomes, which impairs the autophagy process and lead to the formation of large autophagic vacuoles. 11 Severe forms of Pompe disease manifest at birth with cardiomyopathy and muscle weakness, while late-onset forms are characterized by progressive motor dysfunction and respiratory insufficiency. While Pompe disease arises from the deficiency in a unique lysosomal enzyme, Danon disease is caused by mutations in the LAMP2 (lysosomal associated membrane protein 2) gene on the X chromosome, which encodes a glycoprotein essential for lysosomal integrity and functioning. 12 Male patients typically present with a multisystemic phenotype, including cardiomyopathy, muscle weakness, and intellectual disability. Female patients usually exhibit a milder and delayed phenotype, primarily featuring cardiomyopathy. Similar to Pompe disease, glycogen accumulates in skeletal muscle in Danon disease. Consequently, both Danon and Pompe diseases also belong to the large group of lysosomal storage disorders (LSDs), characterized by deficiencies in proteins crucial for lysosomal function and the accumulation of specific materials within lysosomes. 13 Of note, autophagic vacuoles in Danon disease stain positive for (sub-) sarcolemmal components. This unusual nature of the vacuoles inspired Nishino et al. to introduce the term “autophagic vacuoles with sarcolemmal features” (AVSF).14,15 XMEA is another AVM involving primary lysosomal dysfunction that will be discussed in detail in the next parts, in terms of clinical, histological and pathological features.
The other group of AVMs is distinguished by the presence of rimmed vacuoles and includes sporadic inclusion body myositis, GNE myopathy and myofibrillar myopathies.16–18 These myopathies are primarily caused by extra-lysosomal alterations, suggesting that the accumulation of rimmed vacuoles results from secondary lysosomal dysfunction.
XMEA: clinical aspects
The first description of XMEA in 1988 provided clinical reports of five males with X-linked inheritance and weakness of proximal limb muscles. 19 The disease had a very slow progression, with an onset at early childhood. The patients exhibited difficulties in standing up from a sitting position, but they all retained ambulation ability. Creatine kinase (CK) was elevated and no organ systems other than skeletal muscle was affected. In 1995, a second report similarly described five male patients with a juvenile onset, predominant proximal muscle involvement and slow progression of the disease. 20 In the following years, other XMEA cases were reported with equivalent clinical features.7,21–30 Some patients showed high frequency discharges during electromyography without typical symptoms of myotonia. 31 Magnetic resonance also revealed muscle atrophy and degeneration, with fat replacement in several patients.21,22,26,28,31–33 There is generally no sign of cardiac or intellectual involvement in XMEA patients. 34 Notwithstanding, cardiac hypertrophy and vacuolation were reported in one 27-month-old infant, who died from respiratory insufficiency. 35 In parallel, one XMEA patient 7 experienced fatal hepatic cirrhosis at 52 years of age. 36 Liver biopsy revealed vacuolation pattern, suggesting that XMEA pathology is not solely confined to skeletal muscles. The authors proposed that other factors had caused his liver disease, which was ultimately accelerated by VMA21 deficiency. 36 Finally, two XMEA cases were reported with elevated urinary β2 microglobulin, although with no evident renal dysfunction. 21
Strikingly, the age of onset and the degree of severity considerably vary between cases, even in between individuals from the same family. 37 The first reported case of late adult-onset was a 65-year-old patient, with symptoms observed around the age of 55. 38 This patient displayed slowly progressive proximal lower limb weakness and ambulation supported by mechanical assistance. At the opposite extreme of the clinical spectrum, severe forms of XMEA associate with neonatal onset. Symptoms include early ambulation loss, as well as severe proximal weakness of upper and lower limb muscles.32,39–42 A few patients also exhibited impaired ocular movements.23,25,39,41 Five male infants died in the neonatal period due to their inability to breathe and suckle. Two other family members survived through nasogastric feeding and intubation, and displayed typical XMEA features. 43 In another family, two brothers died at 7 and 17, due to multiorgan and heart failure, respectively; their cousin died at birth because of respiratory failure, while their nephew showed extremely severe XMEA. 41 Notably, two twin female patients reported with progressive myopathy may be cases of carrier females clinically affected by XMEA, although there was no genetic confirmation of this diagnostic. 44 The variability observed in XMEA is commonly observed in inherited diseases and may be attributed to genetic or environmental modifiers. However, the small number of XMEA patients reported so far makes it challenging to identify contributing factors. Despite the relatively unspecific clinical presentation of XMEA, the systematic histological characterization of skeletal muscle biopsies, followed by genetic analyses, has greatly accelerated XMEA diagnosis in the last years. Thanks to a prenatal testing approach, a case of XMEA was also diagnosed in a male foetus displaying micrognathia, short limbs, and arthrogryposis. 45 In the mother's previous pregnancy, the foetus showed comparable anomalies and died shortly after birth, which may indicate another case of neonatal lethality due to VMA21 mutations.
XMEA: histopathological features
Histopathological features associated with XMEA are homogeneous between patients. Muscle biopsies typically show myopathic features, with variation in fibre size, muscle fibre splitting, and infrequent necrosis. As in other AVMs, they display autophagic vacuoles in muscle fibres. These cytoplasmic vacuoles generally stain positive for sarcolemma-associated proteins, such as dystrophin, caveolin-3 or dysferlin, and for some basal lamina proteins (e.g., laminin-α2, perlecan), leading to the denomination AVSF, as in Danon disease.46,47 Immunostaining for lysosomal proteins, such as LAMP2, reveals accumulation of lysosomes in XMEA biopsies. Positive staining for LAMP2 distinguishes XMEA from Danon disease, marked by LAMP2 loss, and is used as a key diagnostic element. The presence of small to large vacuoles with single or double membranes in XMEA muscle biopsies can be confirmed by electron microscopy.46,47 In some cases, autophagic vesicles are located close to the sarcolemma, suggesting extrusion of their contents. In these areas, the basal lamina appears duplicated.19,21,23–26,38–40,42,43 Finally, inflammatory markers, such as membrane attack complex with C5b-9 deposition or major histocompatibility complex class I, were reported at the membrane of muscle fibres, as well as within the cytoplasmic vacuoles.20,48–50 Muscle biopsies also show calcium deposition within some fibres and along the sarcolemma. 51
XMEA: identification of the genetic cause
The first description of XMEA families in 1988 and 1995 clearly linked the disease to the X chromosome.19,20 Linkage analysis based on polymorphic DNA markers narrowed down the region co-segregating with the disease phenotype to the segment Xq28.52–54 Follow-up studies on the inheritance of highly polymorphic micro-satellites (or short tandem repeats) present in the Xq28 region in XMEA families further narrowed down the number of putative causative genes.55,56 In 2013, whole-exome sequencing applied to several affected individuals from XMEA families led to the identification of pathogenic mutations in the VMA21 gene, present in the Xq28 region. 7 Since this report, several other patients diagnosed with XMEA have been reported with mutation identified in VMA21.21,22,24,25,28,30,32,33,35,38,39,41,43 A systematic review of all XMEA mutations is shown in Table 1. The table also gives information about the number of patients, disease onset, severity, and clinical manifestations associated with each mutation.
Clinical patterns in XMEA and CDG patients affected by VMA21 mutations.
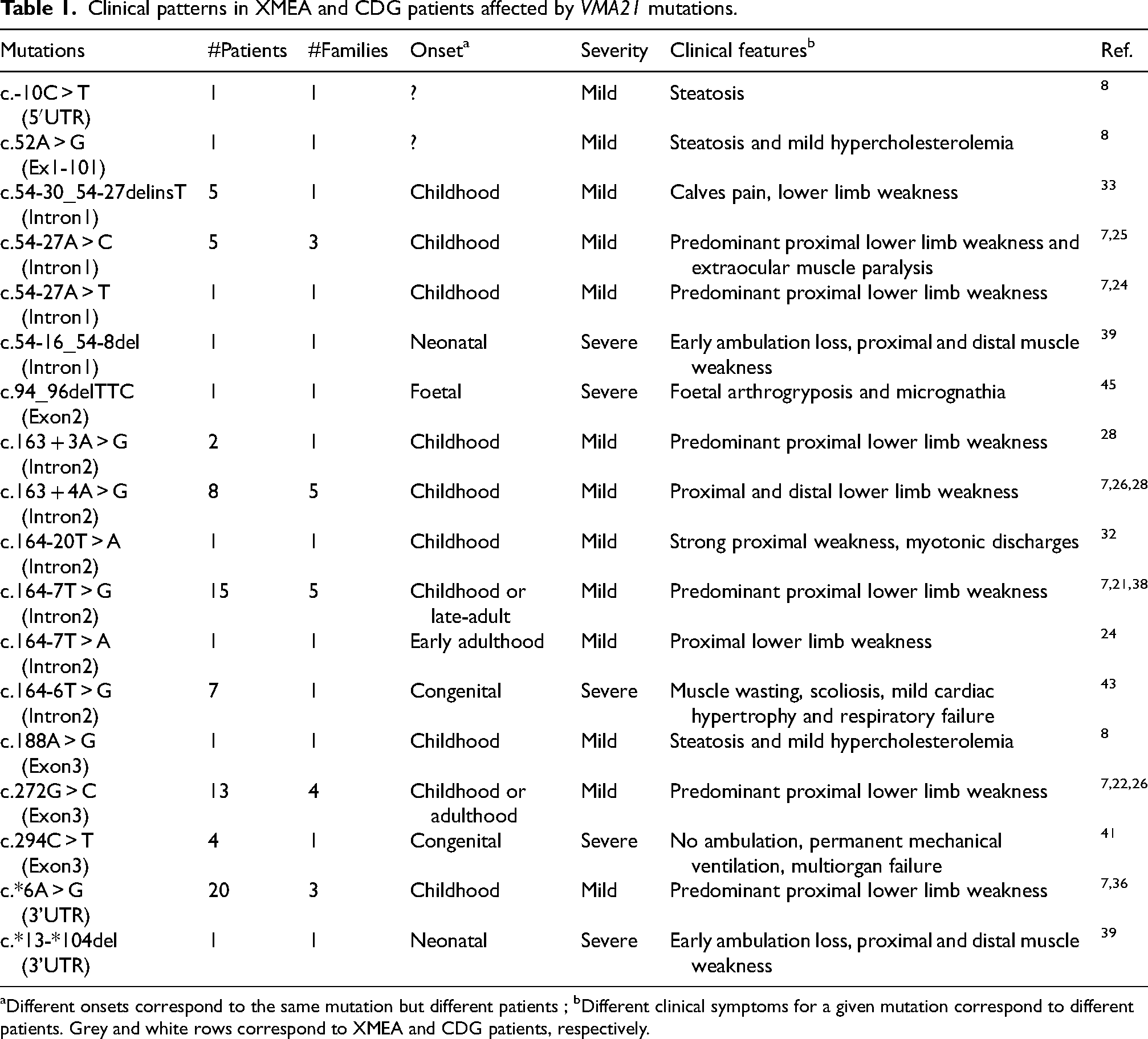
Different onsets correspond to the same mutation but different patients ; bDifferent clinical symptoms for a given mutation correspond to different patients. Grey and white rows correspond to XMEA and CDG patients, respectively.
In the following parts, the nomenclature for the mutations refers to the VMA21-101 transcript (NM_001017980.4), in line with previous case reports. Mutations identified so far are single nucleotide substitutions and microdeletions found mainly in intronic regions of VMA21 (see Figure 1 for the organization of the gene). Classical forms of XMEA were associated with the substitutions c.54-27A > T, c.54-27A > C, c.163 + 3A > G, c.163 + 4A > G, c.164-7T > G, c.164-7T > A and c.272G > C.7,21,22,24,25,28,30 The late onset form of XMEA was also associated with the mutation c.164-7T > G, suggesting involvement of genetic modifiers or environmental factors. 38 Congenital and neonatal cases involved the microdeletions c.54-16_54-8del in the intron 1 and c*.13_*104del in the 3′UTR region, as well as the substitutions c.164-6T > G and c.294C > T.39,41,43 Of note, the mutation c.164-6T > G is located at one nucleotide from the c.164-7T > G mutation associated with classical forms. The patient with cardiac involvement displayed the same mutation (c.164-6T > G), 35 while the XMEA patient with hepatic failure carried a mutation in the 3′UTR region (c.*6A > G). 36 Finally, the prenatal case was associated with the non-frameshift deletion mutation c.94_96delTTC located in a highly conserved region of VMA21. 45 The functional consequences of these mutations will be discussed afterwards.
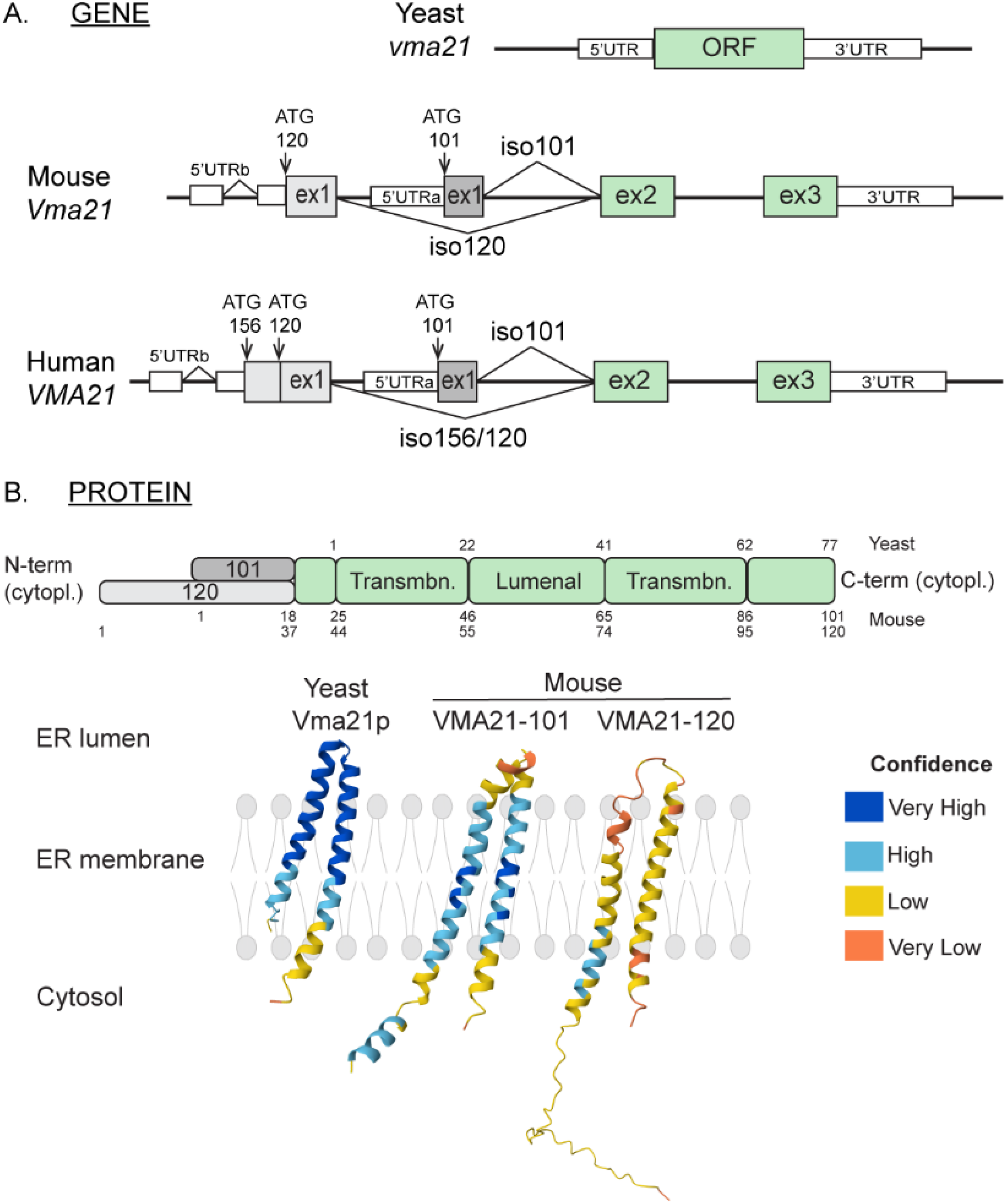
VMA21: from gene to protein. A. Organization of the VMA21 gene in yeast, mouse and human. Exons and untranslated (UTR) regions are represented by grey (exon 1), green (exons 2/3) and white boxes (UTR). Alternative ATG codons are indicated with arrows. B. Representation of VMA21 protein structure in yeast and mouse. Protein topology prediction for VMA21 proteins were obtained with AlphaFold.57,58 Cytopl : cytoplasmic ; Transmbn : transmembrane.
Congenital disorder of glycosylation: expanding the clinical spectrum of VMA21-related disorders
In 2020, mutations in the VMA21 gene were shown to cause a CDG marked by liver disease. 8 CDGs are a group of multisystemic disorders characterized by alterations in the synthesis of glycans.59,60 They are classified based on the type of glycosylation that is altered, e.g., protein N- or O-glycosylation, or glycosphingolipid synthesis. CDGs are caused by an impaired activity and/or expression of enzymes involved in glycosylation, such as glycosidases or glycotransferases, or by an altered activation and/or transport of sugar precursors. The origin of the disease determines the type of glycosylation defect and the clinical pattern observed in the patients. In most CDGs, the liver is clinically affected, contributing to the multisystemic phenotype observed in these patients. 61 Some CDG patients display neurological abnormalities, including mental retardation, seizures, and/or microcephaly. 62 Few patients exhibit cardiac involvement. 63 Screening tests for CDGs are based on the analysis of glycoprotein separation (isoelectric focusing, IEF) based on their isoelectric point. This typically includes transferrin, a predominant glycoprotein in the serum that contains 2 sites for N-glycans eventually terminated with sialic acid. Defects in N-glycan synthesis result in the improper incorporation of sialic acid, which is detectable by IEF in the serum of patients with CDGs.64,65 More than 100 genes have been shown to cause CDGs, with an alteration of the N-glycosylation in the majority of these diseases.
Three male patients with VMA21 mutations have been diagnosed so far with CDG syndrome (Table 1). 8 Abnormal N- and O-glycosylation of plasma proteins was documented for all patients, with the accumulation of markers, such as transferrin or ApoCIII, with low sialylation. The three patients exhibit clinical features ranging from defective bile flow from liver (cholestasis) and fat retention in liver (steatosis) to mild hypercholesterolemia. These symptoms were associated with increased levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) detected at early ages, followed by hypercholesterolemia for two of these patients. 8 There was no sign of myopathy reported for these patients. Of note, levels of ALT/AST tested in one XMEA patient in this study were normal, while cholesterol levels were increased. 8 Ultrastructural analysis of liver biopsies from VMA21-CDG patients revealed mild steatosis and irregularly shaped hepatocytes, without inflammation or fibrosis. Hepatocytes displayed accumulation of lipid droplets within lysosome-related vesicles, multivesicular bodies, as well as dilated Golgi. The mutations identified in VMA21-CDG patients are single nucleotide substitutions detected before the exon 1 (c.-10C > T), in the exon 1 (c.52A > G) and in the exon 3 (c.188A > G). 8 These mutations were not previously reported in XMEA patients, which may explain, at least in part, the clinical heterogeneity observed between patients. Notably, mutations in the genes ATP6AP1, ATP6AP2, TMEM199 and CCDC115, encoding the four other assembly chaperones of the v-ATPase, also cause CDGs with a heterogeneous clinical spectrum including hepatic injury and steatosis.66–70 The corresponding reports do not document myopathic syndrome for these patients. However, some of them presented with psychomotor impairment, hypotonia or muscle weakness, indicating potential skeletal muscle involvement. 66–69
Cancer: a potential role of VMA21?
Follicular lymphoma (FL)
FL represents a common indolent B-cells lymphoma with an estimation of 100.000 cases in the US in 2016. 71 Its incidence strongly varies depending on ethnicity and geography, and it tends to increase with age. Lymphoma usually involves bone marrow and lymph nodes. Symptoms of FL can be subtle and develop gradually, with usually swelling of lymph nodes in different regions of the body, such as the neck, the abdomen, and more rarely, the inguinal region. 72 Around 20% of the patients also experiences fatigue, fever, night sweats and weight loss. Most FL patients carry the somatic chromosome translocation t(14;18) (q32;q21), which results in the constitutive activation of B-cell lymphoma 2 (BCL2) oncogenic protein. 73 In parallel, 10% of FL cases are associated with mutations in the gene RRAGC encoding the small G-protein Ras Related GTP Binding C (RagC), a component of the complex Ragulator that signals amino acid levels and mediates the activation of mammalian Target of Rapamycin Complex 1 (mTORC1). Consistently, increased mTORC1 activity is central in FL and constitutes a potential therapeutic target. 74 More recently, hotspot mutations in the gene ATP6V1B2 encoding a v-ATPase subunit were also associated with LF and mTORC1 hyperactivation. 9 Exome sequencing of FL B-cells led to the identification of other mutated genes, including ATP6AP1, ATP6AP2 and VMA21. 75 Of note, VMA21 mutations were present in cells with concurrent mutations in KMT2D, EP300 and CREBBP, suggesting that VMA21 contributes as a modifier factor in the development or progression of FL. 9 The hotspot mutation identified in VMA21 in FL cells corresponds to the nonsense substitution c.277C > T, which has been reported neither in CDG nor in XMEA. The mutation results in the deletion of nine amino acids in the C-terminal region of VMA21 (p.R93X). 9 Other nonsense mutations (c.263G > A and c.284G > A) affecting the C terminus, as well as several missense mutations (c.128G > A, c.142A > C, c.54-1G > A) were identified in LF cells. Expression of the mutant forms of VMA21 would give a selective advantage to the mutated FL B-cells (see the pathomechanisms discussed below).
VMA21 expression and cancer prognosis
The TIMER2.0 database 76 suggests changes in VMA21 transcript levels in several cancer types, as compared to healthy cells (Figure 2A). In line with this, up-regulation of VMA21 expression was confirmed in lung adenocarcinoma (LUAD) cell lines, as compared to normal human bronchial epithelial cells.77,78 This was linked to increased levels of the long non-coding RNA (lncRNA) ZFPM2-AS1 and of the circular RNA circRNA-0001361 in LUAD cells, which sponged the inhibitory effect of the microRNAs miR-18b-5p and miR-525-5p towards VMA21 transcript.77,78 Similarly, VMA21 transcript and/or protein levels were increased in ovarian cancer cells, melanoma cell lines and cervical cancer cells, which was associated with the up-regulation of other specific lncRNAs.79–81 These changes suggest that VMA21 may play a role in cancer onset and/or progression. This is supported by the correlation observed between VMA21 expression and the prognosis of certain cancers. As such, a positive correlation between VMA21 expression and survival has been reported for colorectal cancer (CRC) . In contrast, high VMA21 expression was linked to poor prognosis in ovarian cancer, melanoma, LUAD (Figure 2B – not significant in the Human Protein Atlas (THPA) database 82 ) and cervical cancer.78–81 High VMA21 expression mediated the effect on cell growth of lncRNAs specifically up-regulated in each cancer type (e.g., LOXL1-AS1 in ovarian cancer).78–81 VMA21 up-regulation may thus be a strong contributor to tumorigenesis in these cancers. Of note, the THPA database indicates that high VMA21 levels rather correlate with favourable patient outcome in cervical cancer (Figure 2C). Finally, the International Cancer Genome Consortium documents the single base substitution c.277C > T (p.R93X) identified in FL with high functional impact in cancers. This variant has been found in one donor with oesophageal cancer and one with colon adenocarcinoma over 332 and 402 donors, respectively. Overall, VMA21 deregulation may associate with either a favourable or unfavourable prognosis depending on the cancer type. This dual effect may be driven by cell type-specific signalling pathways related to cell growth and death, and distinct sensitivities and dependencies to autophagic flux.
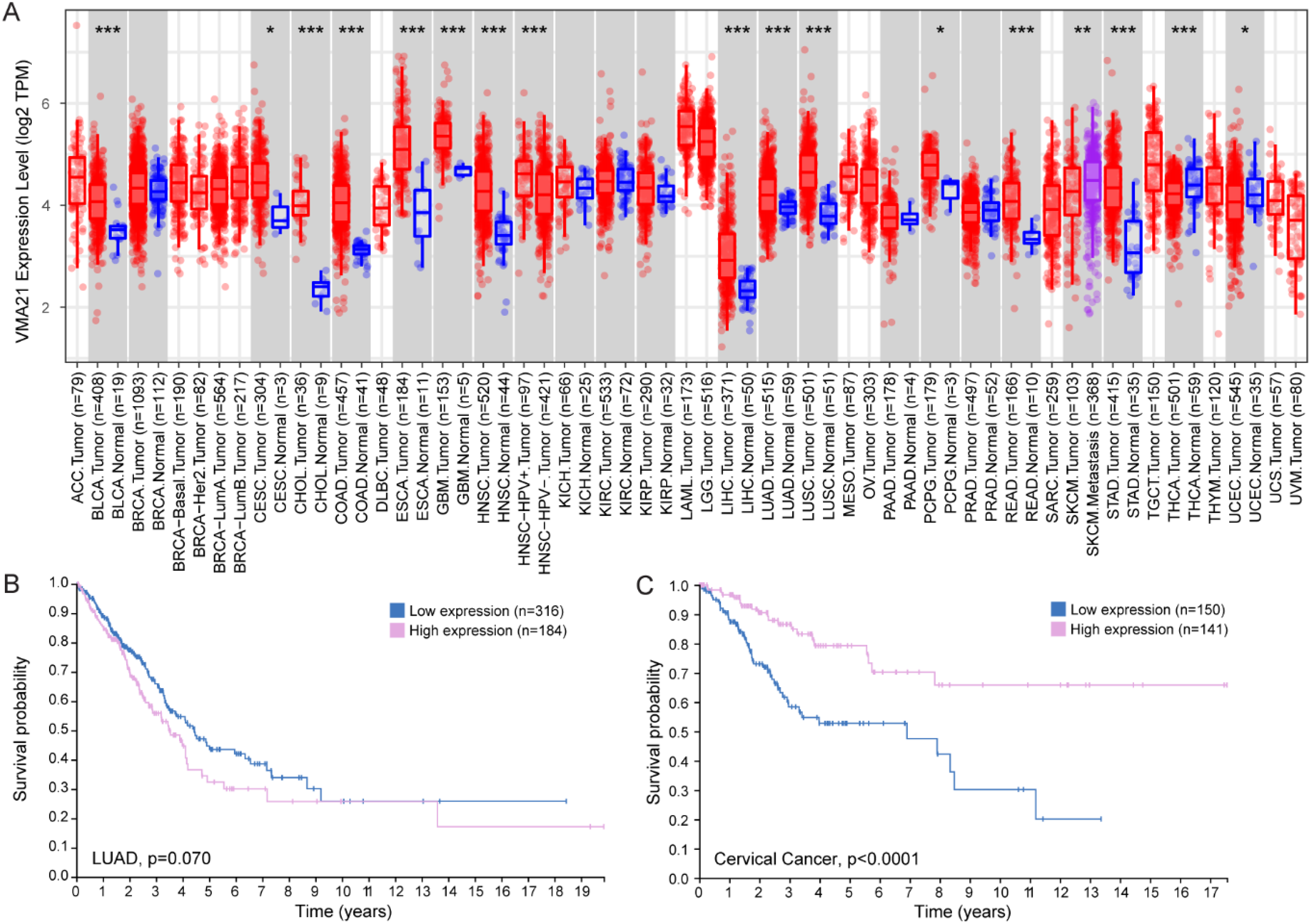
VMA21 in cancer. A. Expression pattern of VMA21 in tumour and normal tissues obtained from TIMER2.0. 76 Distribution of gene expression levels are displayed using box plots. Wilcoxon test, * p < 0.05, ** p < 0.01, *** p < 0.001. ACC : adrenocortical carcinoma; BLCA : bladder urothelial carcinoma; BRCA : breast invasive carcinoma; CESC : cervical and endocervical cancer; CHOL : cholangiocarcinoma; COAD : colon adenocarcinoma; DLBC : lymphoid neoplasm diffuse large B-cell lymphoma; ESCA : oesophageal carcinoma; GMB : glioblastoma multiforme; HNSC : head and neck cancer; KICH : kidney chromophobe; KIRC: kidney renal clear cell carcinoma; KIRP : kidney renal papillary cell carcinoma; LAML : acute myeloid leukaemia; LGG : brain lower grade glioma; LIHC : liver hepatocellular carcinoma; LUAD : lung adenocarcinoma; LUSC : lung squamous cell carcinoma; MESO : mesothelioma; OV : ovarian serous cystadenocarcinoma; PAAD : pancreatic adenocarcinoma; PCPG : pheochromocytoma and paraganglioma; PRAD : prostate adenocarcinoma; READ : rectum adenocarcinoma and stomach; SARC : sarcoma; SKCM : skin cutaneous melanoma; STAD : stomach adenocarcinoma; TGCT : testicular germ cell tumors; THCA : thyroid carcinoma; THYM: thymoma; UCEC : uterus corpus endometrial carcinoma; UC : uterine carcinosarcoma; UVM : uveal melanoma. B, C. Survival analysis of 490 LUAD patients and 291 cervical cancer patients, with low or high VMA21 expression levels. Data are from The Human Protein Atlas. 82
VMA21: from gene to cellular (dys)functions
From gene to protein
VMA21: gene, transcripts and protein isoforms
The Vma21 gene was first discovered in 1994 in Saccharomyces cerevisiae by K. J. Hill and T. H. Stevens from studies screening mutants deficient for v-ATPase function. 83 In yeast, the Vma21 gene is composed of one exon and codes for a small protein of 77 amino acids. Remarkably, Vma21 null mutants exhibit defective vacuole acidification and undetectable v-ATPase activity, together with impaired growth. 83 In mammals, the VMA21 gene is located on the X chromosome (Xq28 region in human) and the coding sequence is composed of three exons. Different VMA21 transcripts are predicted in silico in human, as well as in other mammals, such as mice. These transcripts arise from alternative transcription start sites (TSS) and share exons 2 and 3, but not exon 1 (Figure 1A). We recently demonstrated that two main transcripts are expressed in humans and mice: one short transcript (VMA21-101) reported in prior studies and one longer transcript (VMA21-120). 84 Notably, a third transcript, VMA21-156, was detected in human but not in mice. The TSS of VMA21-120/156 are located upstream the TSS of VMA21-101 (Figure 1A).
In yeast, Vma21 encodes Vma21p of 8.5 kDa. In human, the alternative transcripts encode a 101-, 120- and 156-amino acid long proteins, of 11.5, 13.5 and 18 kDa, which differ in their N-terminal regions 84 (Figure 1B). In both human and mouse, VMA21-101 expression is detected in most tissues. In contrast, VMA21-120 expression is restricted to skeletal muscles and is higher during muscle development and regeneration in mice. Consistently, VMA21-120 expression strongly increases after inducing muscle cell differentiation in vitro, before the fusion of committed muscle precursors. 84 Based on RNAseq data, VMA21-120/156 also seem to be specifically expressed in the muscle lineage in human. 84 Low levels of these isoforms were found in heart. Protein sequence alignment revealed a strong conservation throughout species for the region shared by the three isoforms (central and C-term). The N-terminal region of human VMA21-101 shows high similarities with homologous proteins found in rodents, fish or worm, but not in S. cerevisiae. 84 Notwithstanding, expression of human VMA21-101 was sufficient to reverse growth defects in yeast vma21 null mutants. 7 Homologs of VMA21-120 were identified only in mammals, and only in primates for VMA21-156. 84 VMA21 isoforms have also been described in plants: AtVMA21a and AtVMA21b share around 28% protein similarity with Vma21p, and were both sufficient to compensate for Vma21p depletion in yeast. 85
From VMA21 mutations to clinical spectrum
Mutations found in patients affected by XMEA or CDG are distributed across the entire VMA21 gene (Figure 3), with the exception of VMA21-120 exon 1 and surrounding regions, which have not been, to our knowledge, included in the exome strategy sequencing so far. All VMA21 mutations are inherited in a recessive X-linked manner and correspond to microdeletions or single base substitutions. The outcome of VMA21 mutations is illustrated in Table 2. As discussed above, most mutations associated with XMEA are in intronic regions, affecting donor or acceptor splicing sites. In particular, Ramachandran and al. (2013) initially described 4 substitutions in non-coding regions with distinct effects on RNA splicing: the mutations c.54-27A > T and c.54-27A > C result in loss of the adenine critical for the splice branch point of intron 1; c.163 + 4A > G eliminates the adenine in the fourth position after exon 2, impairing the binding of the U1 snRNA during splicing; and c.164-7T > G before the exon 3 is proposed to decrease U2AF splice factor binding efficiency. 7 Notably, the mutation c.272G > C in the coding sequence (p.Gly91Ala) and the silent mutation c.294C > T (p.Gly98=) would also affect VMA21 splicing. 41 In parallel, mutations reported in the 3′UTR region (c.*6A > G or c.*13_*104del) in proximity to the stop codon, were associated with transcript destabilization.7,39,84
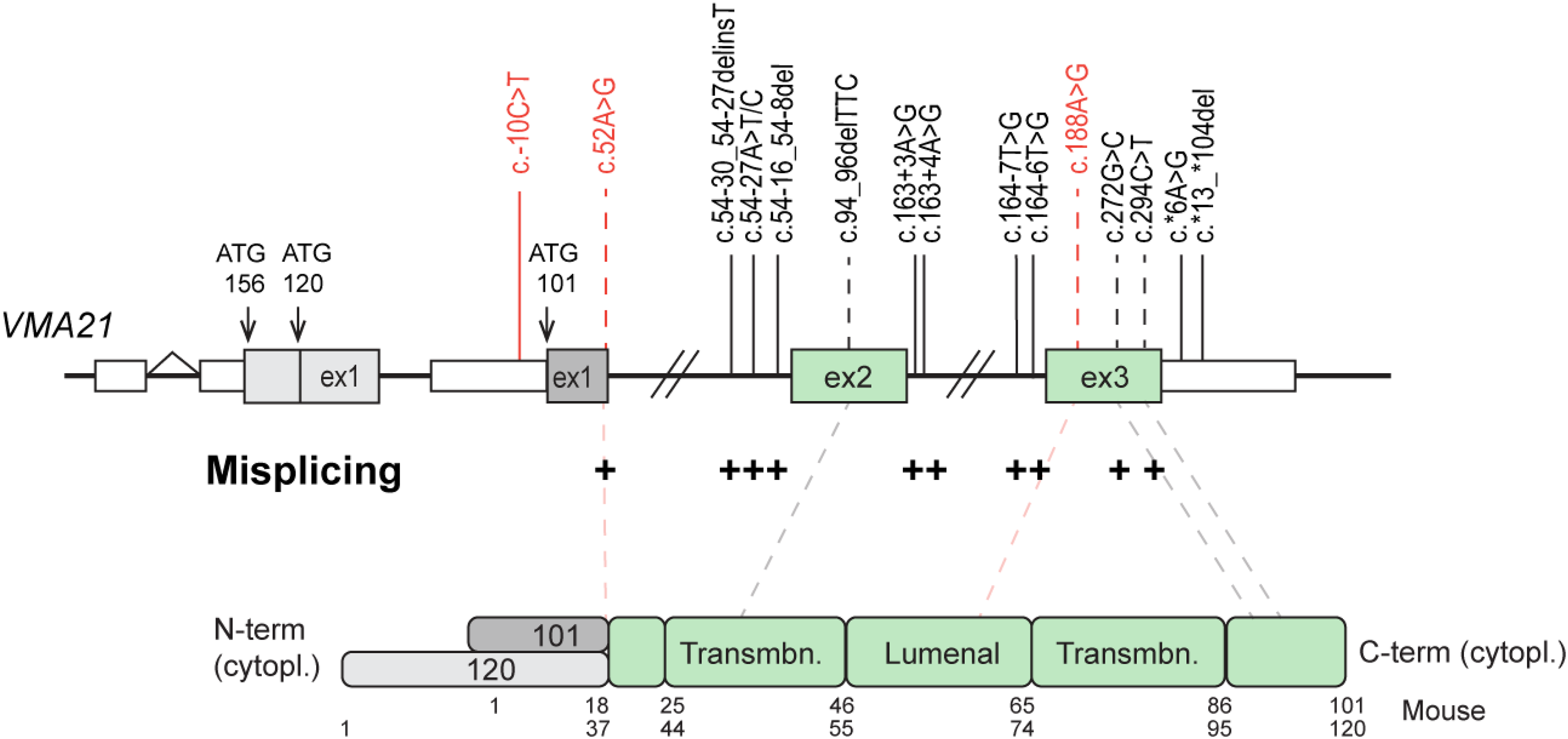
Spectrum of VMA21 mutations identified in CDG and XMEA. Mutations identified in XMEA and CDG patients are in black and red, respectively. The position of the TSS of the three VMA21 isoforms are shown with arrows on the gene (top). Exons and UTR regions are shown with boxes; introns are depicted with bridging gaps and lines. Amino acids numbers are indicated below the protein domains (down). Mutations in coding region are positioned at the protein with dotted lines. Mutations affecting splicing are indicated with + . Cytopl: cytoplasmic; Transmbn: transmembrane.
Effect of VMA21 mutations on mRNA and protein levels of VMA21.
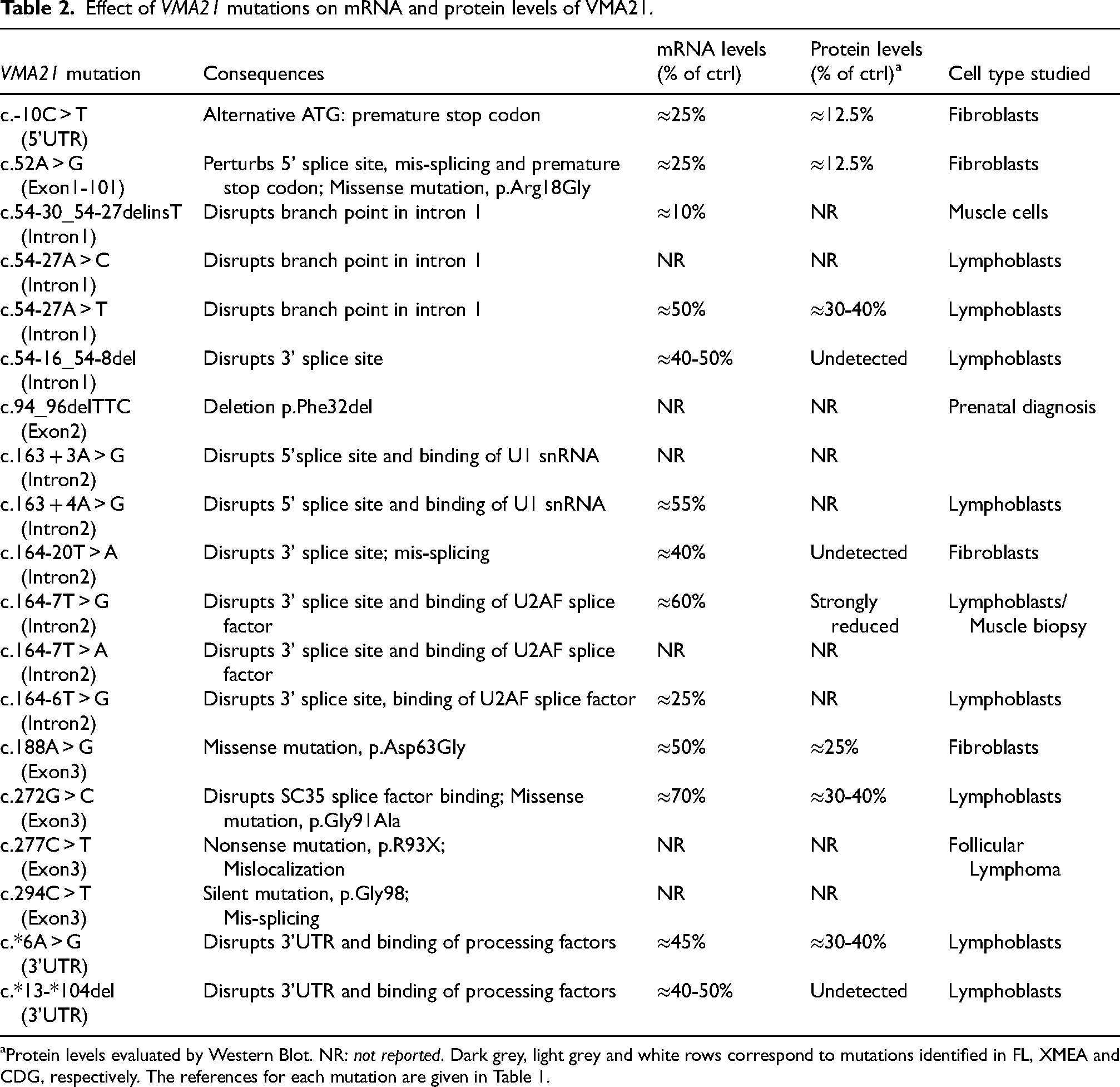
Protein levels evaluated by Western Blot. NR: not reported. Dark grey, light grey and white rows correspond to mutations identified in FL, XMEA and CDG, respectively. The references for each mutation are given in Table 1.
The severity of XMEA has been linked to the residual levels of VMA21. Indeed, severe cases showed drastic reduction in VMA21 transcript or protein levels in lymphoblasts or fibroblasts, while 30-60% remaining expression levels were reported for other XMEA mutations (Table 2). However, VMA21 protein levels have not been evaluated for all mutations. Moreover, recent studies reported lower transcript and/or protein levels when using muscle cells.33,84 Hence, the consequences of each mutation on VMA21 expression, especially focusing on both VMA21 isoforms, should be evaluated in muscle cells, to get a more representative view of VMA21 deficiency in XMEA patients, and of the correlation between VMA21 levels and disease severity.
Analysis of VMA21 mutations in CDG patients by Cannata Serio et al. (2020) also provided information about their molecular consequences. 8 The mutation c.-10C > T in the 5′ UTR of VMA21-101 produces a new ATG initiation codon located 11 bp upstream of VMA21-101 exon 1, which results in a premature stop codon in between nucleotides 26–28 of VMA21 coding sequence. Similarly, the mutation c.52A > G in exon 1 of VMA21-101 leads to the formation of a predominant alternative splice variant and to a premature stop codon immediately at the end of the exon 1. Fibroblasts from CDG patients carrying these mutations expressed only 10-20% remaining VMA21 protein levels. Finally, the mutation c.188A > G in exon 3 results in the substitution of an asparagine to glycine (p.Asp63Gly) in the luminal domain, with around 30% reduction in protein levels compared to control. 8 Whether the function of the mutated protein is preserved remains unknown.
Overall, it is difficult to explain the distinct clinical spectrum between XMEA and CDG on the basis of the remaining expression levels of VMA21. In most studies, only the levels of VMA21-101 have been evaluated in cells from patients. The microdeletion c.54-30_54-27delinsT in intron 1, reported in a Brazilian XMEA patient, strongly alters the expression of both VMA21 isoforms.33,84 The mutation led to an alternative VMA21-120 transcript containing a small sequence of intron 1, which resulted in a premature stop codon; the corresponding protein was undetected in patient cells. Similarly, the mutation c*13_*104del, reported in an Italian XMEA patient, reduces by 70-80% VMA21-101 and VMA21-120 transcript levels in patient myotubes.39,84 The specific expression of VMA21-120 in skeletal muscle, as well as the localization of VMA21 mutations in XMEA vs. CDG patients, suggest that the loss of VMA21-120 may contribute to XMEA pathogenesis. Mutations affecting VMA21-120 expression may primarily trigger muscle phenotypes. Consistently, all XMEA patients carry mutations close to or in exon 2, exon 3 or in the 3′UTR, which are common sequences of VMA21-101 and -120 transcripts. In contrast, two of the CDG mutations were found in or close from VMA21-101 exon 1, which may preserve VMA21-120 expression and spare skeletal muscles in CDG patients (Figure 3). However, why the loss of VMA21-101 in XMEA patients does not result in liver or other non-muscle tissue alterations is still unknown. Further investigation on the expression levels of VMA21-120 in XMEA and CDG, as well as inclusion of VMA21-120 exon 1 in exome sequencing strategy, will be decisive in better understanding VMA21-associated disorders.
VMA21: properties of the chaperone protein
Sequence analysis and Alpha Fold57,58 predict that VMA21 is a transmembrane protein with both N- and C-terminal regions exposed to the cytosol 83 (Figure 1B). CryoEM in yeast confirmed the α helical hairpin topology of Vma21p within the membrane, with the two side chains in the cytosol. 86 AlphaFold predictions suggest differences between VMA21 isoforms in their respective N-terminal domain. The N-terminal region of VMA21-120 consists of 37 residues, which would form a flexible domain in the cytoplasm. In contrast, VMA21-101 would feature a short helical domain (residues 2-11), followed by a 3-residue linker (residues 12-14) connected to the first transmembrane helix (Figure 1B). The different length and conformation of the N-terminal domains of VMA21 isoforms may impact the structural dynamics, the interactions with other proteins, and thereby the function of each isoform.
The topology of Vma21p/VMA21 is consistent with its localization in the membrane of organelles. Most studies have revealed an accumulation of the protein in the ER in yeast83,87 and in mammals.7,84 The protein was also found in the ER-Golgi intermediate compartment (ERGIC) in muscle cells and lymphoblasts.7,84 In contrast, VMA21 was undetectable in lysosomes or in the Golgi. In yeast, Vma21p contains a carboxy-terminal di-lysine motif (KKXX) that is critical for its ER retention. Mutagenesis of these lysine residues resulted in Vma21p delivery to the yeast vacuolar membrane. 83 The di-lysine motif is conserved in plants, but not in animals. The alternative motif contains a single lysine and an acidic aspartate residue (XKXD), which would ensure the retention and traffic back of VMA21 to the ER.7,9 Indeed, mutation of the KQD motif of the human VMA21 led to its mislocalization to lysosomes, providing evidence that the KQD residues serve as an atypical ER retrieval signal. 9
VMA21: an assembly chaperone of the v-ATPase
Functions and organization of the v-ATPase
v-ATPases are large protein complexes found in all eukaryotes, which form membranous proton pumps, essential for the acidification of organelles and intracellular vesicles. This acidification sustains specific functions in sub-cellular compartments. 88 In lysosomes, the low pH (pH 4-5) confers an optimal environment for the activity of degradative enzymes. In Golgi, major post-translational modifications and protein sorting process efficiently at a pH of 6.2-7. In specific cell types, v-ATPases are also found at the plasma membrane, where they contribute to acidify the extracellular environment. 89
The yeast v-ATPase is a large complex consisting of a cytoplasmic V1 domain formed of 16 sub-units (A3,B3,C,D,E3,F,G3,H) responsible for ATP hydrolysis, and of a transmembrane V0 domain responsible for proton translocation, composed of the subunits a,c8,c’,c’’,d,e,f and the recently discovered Voa1p90,91 (see Figure 4). The sub-units c, c’ and c” form the c-ring of the V0 domain. Mammalian v-ATPases closely resemble yeast v-ATPase organization. However, the sub-units ATP6AP1 and RNAseK replace Voa1p and the sub-unit f in the V0 domain. Moreover, the c8c’c’’-ring is replaced by the sub-units c9c”, with the additional sub-unit ATP6AP2.92–94 Finally, CryoEM of the v-ATPase in porcine kidney and rat brain suggest that a small proportion of V1 domain may contain an additional C subunit.93,95
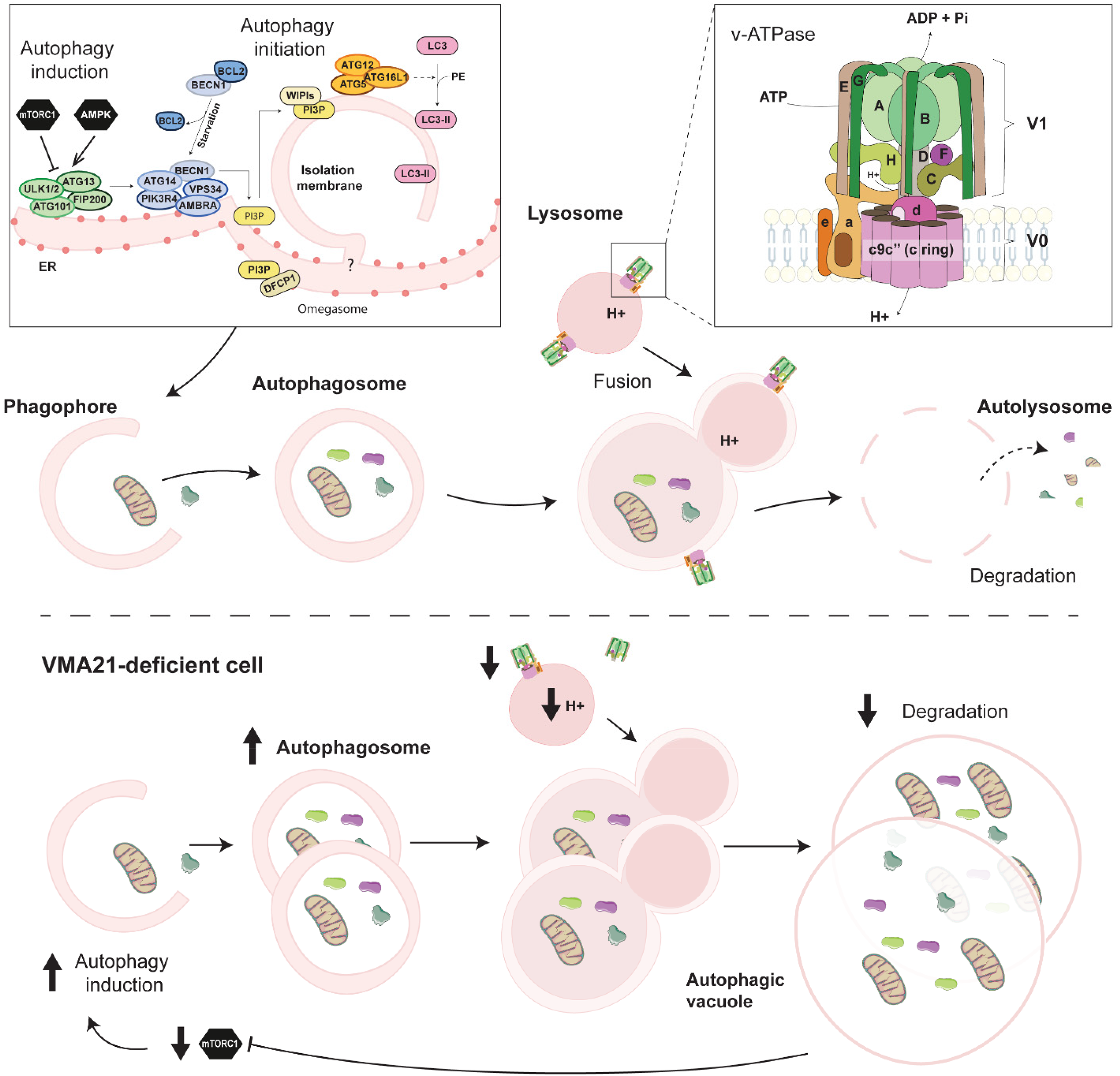
Autophagic flux and autophagy impairment in VMA21-deficient cells. Overview of the autophagy process and of the proteins involved in mammals. Autophagy induction depends on the activation state of mTORC1 and AMPK, which regulate the Ulk1 complex. Autophagic vesicles engulf large cytoplasmic parts, including organelles, which are degraded after fusion with lysosomes and recycled. Defective degradation steps in VMA21-deficient cells lead to the formation of AVSF.
Interestingly, only the sub-unit V0a exists in two isoforms in yeast. In contrast, mammalian v-ATPases have multiple isoforms of each sub-unit, which are expressed in specific tissues, cells or organelles. 88 For example, the a2 isoform of the subunit V0a is included in v-ATPases present in the Golgi and early endosomes, but not in lysosomes. Mutations in ATP6V0A2, encoding this specific isoform, cause autosomal recessive cutis laxa type 2 (ARCL2A) characterized by developmental delay, skin hyperelasticity and neurologic abnormalities, and associated with defects in glycosylation and endocytosis.96,97 Similarly, mutations in ATP6V0A3, ATP6V0A4 or ATP6V1B1, encoding the osteoclast-specific V0a3 subunit, and the isoforms V0a4 and V1B1 predominantly expressed in kidney and inner ear, cause osteopetrosis, renal tubular acidosis and/or deafness.98–100
Mechanisms of v-ATPase assembly
Biochemical and genetic analysis in yeast have revealed that Vma21p and two additional chaperone proteins, Vma12p and Vma22p, are required for the assembly of the v-ATPase enzyme, but not included in the fully assembled complex.90,101–103 Vma12p and Vma22p are homologous to mammalian TMEM199 and CCDC115. High-resolution cryoEM provided detailed information on how these chaperones cooperate for the assembly of the V0 domain in the ER, before it binds V1 in the Golgi. 104 In their recent study, Wang et al. described the structure of V0:Vma12p-Vma22p and V0:Vma21p complexes. 86 Vma12p and Vma22p initially bind the subunit V0d and the c ring. The subsequent interaction between Vma12p and the subunit V0a facilitates the recruitment of V0e and V0f subunits, thereby completing the assembly of V0. Vma22p occupies the binding site of the subunit V1D on the sub-unit V0d, preventing premature interaction between V1 and V0 domains. The Vma12/22p complex is released when V0 undergoes a conformational change to a lower energy state. At this stage, V0 is escorted by Vma21p to the Golgi apparatus. 86 Vma21p binds to different sites around the c-ring of the immature V0 domain: Vma21p preferentially binds V0c2 and V0c3, although multiple copies of Vma21p may bind around the c-ring at low density. On top of that, Wang et al. also provided evidence of two yet-unidentified proteins, YAR027W and YAR028W, that interact with the c subunits and with Vma21p. 86 Although the precise location of these proteins in the c complex remains unclear, this suggests that Vma21p activity may depend on the interactions with other binding partners. Upon reaching the Golgi apparatus, the V0 complex binds the V1 domain, releasing Vma21p. Due to its retrieval signal, Vma21p cycles back to ER, while the v-ATPase is transported to its final destination.
The role of VMA21 in chaperoning the assembly of V0 domain has also been reported in mammals. In human cells, VMA21 was shown to interact with v-ATPase V0c’’ subunit. 7 Esmail et al. (2018) also showed by co-immunoprecipitation that VMA21 binds glycosylated, but not unglycosylated, V0a subunit. 105 The authors conclude that the oligosaccharide moieties may function as a recognition signal for VMA21, which may retain unglycosylated subunits in the ER and prevent their incorporation into V0 domain. More recently, we showed that both VMA21 isoforms interact with the V0 domain through binding with the c subunit in mouse muscle cells. 84 Our study also revealed direct or indirect interaction between the two VMA21 isoforms, suggesting synergistic or competing effect towards V0 assembly. Finally, in line with a role of VMA21 in chaperoning v-ATPases, VMA21 deficiency in cells from XMEA patients reduced v-ATPase activity, potentially as a result of diminished v-ATPase levels. V1E subunit accumulated in the cytosolic fraction, suggesting decreased formation of the V0/V1 complex. 7
VMA21 circRNA
Apart from VMA21 proteins, recent evidences emerged regarding the existence of circRNAs encoded by VMA21. CircRNAs are a specific type of non-coding RNA, which are single-stranded and covalently closed biomolecules without 5′ or 3′ ends. They are evolutionarily conserved across species, from viruses to mammals, and can arise from exons (exonic circRNA) or introns (intronic circRNA). 106 CircRNAs can act as protein decoys and scaffold for protein interaction. In addition, circRNAs harbour multiple binding sites for miRNAs, thereby serving as inhibitors of miRNAs. 107 circRNAs unique expression signature has been involved in cancer, 108 autoimmune 109 and cardiovascular diseases. 110
In recent years, an increasing number of studies pointed to the involvement of circVMA21 in inflammatory conditions, through inhibition of specific miRNAs. In a rat model of intervertebral disc degeneration, circVMA21 targeted miR-200c/XIAP (X-Linked Inhibitor Of Apoptosis) pathway and alleviated inflammation and apoptosis. 111 Similarly, circVMA21 reduced inflammation in different models of sepsis and kidney injury, by sponging miRNAs, such as miR-497-5p and miR-7-5p.112,113,114,115,116 Recent findings also revealed chondroprotective effects of circVMA21 via acting on miR-495-3p/FBWX7 (F-Box And WD Repeat Domain Containing 7) 117 and miR-103, 118 providing valuable directions for the treatment of osteoarthritis. Overall, decreased expression of circVMA21 associated with increased inflammation in all pathologies, while its overexpression relieved inflammatory injury, pointing to circVMA21 as a promising therapeutic target. It is noteworthy that current evidence also suggests that circRNAs can code uncharacterized proteins with high sequence homology compared to mRNA-encoded isoforms. This makes them attractive for a competitive binding to partners of cognate proteins, in what is known as the “protein bait hypothesis”. 119 Whether circVMA21 perturbs the activity of VMA21 proteins and/or participates in the progression of XMEA, VMA21-CDG or lymphoma still needs to be studied.
VMA21-associated pathomechanisms: from cellular functions to pathogenesis
Autophagy impairment
Regulation of the autophagic flux
Autophagy is an intracellular degradation process in which cytoplasmic materials and organelles are degraded in lysosomes. Among other types of autophagy, macroautophagy (hereafter simply referred to as autophagy) ensures basal elimination and recycling of defective organelles or misfolded proteins. Its induction increases in stress conditions conferred by oxygen, nutrient or growth factor deprivation.120,121 Autophagy involves the formation of a phagophore that engulfs targets and elongates to form a double-membrane vesicle, called autophagosome. 122 The fusion of autophagosome with lysosomes leads to autolysosome, in which lysosomal enzymes degrade the autophagic content (Figure 4). The autophagic flux is hence determined both by the state of autophagy induction and the efficiency of the degradation steps.120,121
Proteins encoded by autophagy-related genes (Atg), as well as other key factors, such as the cargo receptor p62, tightly orchestrate autophagy. The extent of autophagosome formation is mainly determined by mTORC1 and 5′ AMP-activated protein kinase (AMPK), which inhibits and increases autophagy induction, respectively (Figure 4). In growth conditions, activation of mTORC1 leads to phosphorylation and inhibition of the serine/threonine kinase Ulk1, involved in autophagy initiation.123,124 Contrarily, under starvation or other stress conditions, mTORC1 inhibition releases Ulk1 activity and triggers autophagy induction. In these conditions, AMPK activation contributes to the inhibition of Raptor, one of the components of mTORC1, and activates Ulk1 by phosphorylating serines 317 and 777.125–127 Although precise evaluation of the autophagic flux represents a limitation in humans, there is increasing evidence that its perturbation contributes to a large spectrum of diseases. 121 In skeletal muscle, insufficient autophagic flux, conferred by blockade of the induction or degradation steps, as well as excessive autophagic flux, trigger loss of muscle homeostasis, muscle atrophy, and potentially muscle degeneration.128–130 Autophagy impairment emerged as one of the main contributor of many myopathies, 10 and several evidence suggest that it is central in the pathomechanisms leading to VMA21-associated diseases.
Evidence of autophagic dysregulation caused by VMA21 deficiency
Acidification of the lysosomal lumen is decisive for the activity of lysosomal enzymes, and thereby for the degradation steps of autophagy. Nakamura and al. first demonstrated the importance of v-ATPase-mediated acidification in autophagic degradation in yeast. 131 They showed that vma mutants (i.e., deficient for v-ATPase) accumulate autophagic bodies in the vacuole resulting from defective degradation. In particular, yeast lacking Vma21p do not contain acidified vacuoles and exhibit similar phenotypes as other vma mutants. 83 In line with this, inhibition of lysosomal function with bafilomycin A, a v-ATPase inhibitor, prevented luminal acidification and lysosomal enzyme activity, with a robust block of autophagic flux in Drosophila. 132
The first evidence of autophagy impairment in XMEA is the accumulation of autophagic vacuoles in muscle biopsies from patients. These vacuoles stain positive for the lysosomal marker LAMP2, and for the autophagic markers p62 and LC3, which confirm their autophagic nature.32,33,40,46 Electron microscopy further confirmed the accumulation of autophagosomes and vacuoles containing cell debris in skeletal muscle, as well as in fibroblasts, from XMEA patients.19,21,23–26,38–40,42,43 In lymphoblasts from XMEA patients, Ramachandran et al. (2013) evaluated the remaining v-ATPase activity at 12–22% of control cells, which was accompanied by higher lysosomal pH (pH 5.2 vs. 4.7). 7 In parallel, the elevation of LC3II and p62 levels in patient's fibroblasts and lymphoblasts was consistent with a blockade in the degradation steps of autophagy and reduced autophagic flux. 7 Interestingly, patient cells also displayed major increase in Beclin-1 and Vps34 levels, as well as reduced phosphorylation of mTORC1 targets. Consistently, they uncovered a reduction in intracellular free amino acids that may arise from the partial block of autophagy. The authors hence suggested that the primary autophagy blockade, by reducing amino acid levels, triggers mTORC1 inhibition, and thereby increases autophagy induction. This feedback may exacerbate the accumulation of autophagic vesicles and vacuoles in cells from XMEA patients (Figure 4).
Cannata Serio et al. (2020) also found v-ATPase dysfunction in fibroblasts from VMA21-CDG patients, as well as reduced lysosomal acidification. 8 Increased p62 and LC3 levels suggested autophagic blockade in fibroblasts from VMA21-CDG patients, similar to that observed in XMEA patients. Notably, autophagic defects were associated with an accumulation of lysosome-associated structures and an alteration of lysosomal morphology in VMA21-CDG fibroblasts. Electron microscopy confirmed that autolysosomes accumulate in Kupffer cells and hepatocytes in a liver biopsy of a VMA21-CDG patient. 8
Interestingly, VMA21 variants associated with FL also led to autophagy blockade, 9 similar to XMEA and VMA21-CDG. Electron microscopy showed an accumulation of autolysosomes in LF cells mutated in VMA21. As shown in XMEA cells, there was also a compensatory increase in autophagy induction in these cells. The role of autophagy in cancer cells is controversial. Many studies suggest that autophagy may prevent malignant transformation, whereas other studies have shown that autophagy is involved in cancer development, metastasis and cancer-drug resistance.133,134 Wang et al. suggested that increased autophagic induction in VMA21-mutated cells may be a key factor determining survival for tumour cells, which may be pharmacologically targeted in therapeutic strategies. 75 Accordingly, targeting HEK293T cells harbouring VMA21 mutations with a drug inhibiting autophagy induction effectively killed these cells, pointing to novel therapeutic strategies for FL associated with aberrant v-ATPase activity. However, these strategies should be adapted depending on the tissue and cancer type. Indeed, as mentioned above, VMA21 may function both as a tumour suppressor and tumour promotor. Whether the role of VMA21 in modulating tumorigenesis only relies on its effect on the autophagic flux remains unclear.
Therapeutic strategies
Since reduced levels of VMA21 in XMEA and CDG cells blocks autophagic degradation and in turn increases autophagy induction, it becomes challenging to propose a straightforward approach based on autophagy modulation as a therapeutic strategy. Ideally, this strategy would limit autophagy induction and restore lysosomal pH with acidifying agents, in order to prevent accumulation of autophagic vesicles. Lysosomal re-acidification has been proposed as a potent approach for LSDs. 13 Biodegradable acidic nanoparticles restored lysosomal acidity and autophagy in mouse models of non-alcoholic fatty liver disease (NAFLD). 135 These nanoparticles remain intact and inactive at plasma pH, while they get degraded around pH 6, which may correspond to the luminal pH of lysosomes in the case of impaired acidification. Acidic nanoparticles also improved lysosomal function and the phenotype of mouse models of neurodegenerative disorders, such as Parkinson's or Alzheimer's diseases (AD).136,137 Interestingly, presenilin1 (PS1) involved in AD interacts with the subunit V0a1 of v-ATPase, and thereby regulates its delivery to lysosomes. 138 PS1 deficiency disrupted this interaction, reduced accumulation of functional v-ATPase in lysosomes, and impaired lysosomal function in the brain of AD mouse models. However, these defects were not reproduced in cells or mouse brain deficient for PS1/2. 139 Modulating v-ATPase activity with specific compounds, such as dendrobium alkaloids, enhanced lysosomal acidification and improved learning function in β-amyloid precursor protein (APP)/PS1 double transgenic mice. 140 In line with this, targeting V1A subunit with the small molecule EN6 effectively cleared toxic Tar-DNA binding protein 43 (TDP-43) aggregates in a cell line model of amyotrophic lateral sclerosis (ALS) pathogenesis. EN6-mediated clearance of protein aggregates involved mTORC1 inhibition, which subsequently enhanced the nuclear translocation of transcription factor EB (TFEB), increased the expression of genes encoding v-ATPase subunits, and ultimately improved lysosomal acidification. 141 Similarly, overexpressing TFEB in muscle cells or in mouse models for Pompe disease reduced lysosomal size and glycogen accumulation, and enhanced cellular clearance by promoting lysosomal exocytosis. 142 Of note, inhibition of autophagy induction by targeting Atg5 / Atg7 increased the benefits of the enzyme replacement strategy in mouse models of Pompe disease. 143 These recent pharmacological developments open promising perspectives for the restoration of lysosomal acidification in skeletal muscle and/or liver in patients suffering from XMEA and CDG. However, it remains unknown whether the normalization of lysosomal function and autophagy will be sufficient to reverse muscle/liver phenotypes, as XMEA/CDG pathogenesis may involve other VMA21-dependent processes.
Glycosylation defects
N- and O-glycosylations are prevalent post-translation modifications occurring within the Golgi apparatus.144,145 N-glycosylation generally corresponds to the attachment of a glycan to a nitrogen of an asparagine residue within the consensus sequence Asn-X-Ser/Thr, with X representing any amino acids except proline. In O-glycosylation, the glycan is attached to the hydroxyl oxygen of a serine or a threonine residue. Glycosylation has been identified on more than 3000 proteins. 146 As mentioned above, CDGs are typically severe multisystemic disorders, involving different glycosylation defects.
CDG patients with VMA21 mutations showed N- and O-glycosylation abnormalities with marked accumulation of truncated glycans lacking sialic acid and galactose. 8 Interestingly, mutations in genes encoding the other chaperones of the v-ATPase, CCDC115 and TMEM199, or the associated proteins ATP6AP1/2, led to similar combined N- and O-glycosylation defects and to hepatopathy.66,67,69,70 Mutations in the gene encoding v-ATPase V0a2 subunit have also been linked to impaired glycosylation, which may be key in the pathogenesis of cutis laxa type II syndrome. 96 The glycosylation defects reported in these disorders are typically occurring in the trans Golgi. Increase in trans Golgi pH, caused by v-ATPase deficiency, likely alters the activity of enzymes involved in the last glycosylation steps. In line with this, neutralization of Golgi pH with bafilomycin A impaired glycosylation and resulted in the delocalization of glycosylation enzymes. 147 Notably, mutations in ATP6V0A2 altered the retrograde translocation and fusion of membranes from Golgi to ER in fibroblasts. 96 This defective trafficking may also contribute to glycosylation perturbations in patients. However, the morphology of Golgi and ER organelles was preserved upon bafilomycin A treatment, suggesting that v-ATPase activity is not essential for Golgi/ER network regulation. 147 Similarly, cells with mutations in ATP6AP1/2, CCDC115 or ATP6V0A2 showed normal Golgi structure.68–70,96 In contrast, VMA21 and TMEM199 deficiency led to dilated Golgi in hepatocytes.8,67 Hence, genetic deficiency in v-ATPase assembly factors is associated with glycosylation defects, but whether these defects arise from impaired Golgi acidification has not been thoroughly investigated. It is noteworthy that protein glycosylation was measured in three different XMEA patients and found to be normal.8,148
Lipid metabolism dysregulation
Several evidences suggest that VMA21-associated diseases involve dysregulation of lipid metabolism. Liver plays central functions in the capture, storage, transformation and release of lipids. Free fatty acids (FFAs) captured by hepatocytes are used in β-oxidation, or alternatively converted into triglycerides (TG) and stored in lipid droplets (LD) or released in the circulation as very low-density lipoproteins (VLDL). Cholesterol from blood, included in low-density lipoprotein (LDL), enters in hepatocytes, where it can be stored in LDs as cholesteryl esters. A tight regulation of cholesterol storage is required to maintain homeostatic levels of free, potentially toxic, cholesterol in cells. 148 In this context, lysosomes play a major role in regulating the intracellular trafficking and hydrolysis of cholesterol.149,150 Endocytosed cholesterol-LDL are delivered to lysosomes, where lysosomal acid lipases hydrolyse esterified cholesterol into free cholesterol. Notably, the dissociation of cholesterol-LDL from LDL receptors after internalization requires proper acidification of the endosomes. From the lysosomal lumen, cholesterol is then conveyed to the lysosomal membrane through interaction with different proteins, including Niemann-Pick C (NPC) 1/2 proteins, as well as LAMP2 and lysosomal integral membrane protein (LIMP2).151–153 These interactions ensure the transport of cholesterol to other organelles, such as the ER, as well as to the plasma membrane. In the NPC lysosomal storage disease, NPC1/2 deficiency leads to the accumulation of cholesterol and other lipids in lysosomes and endosomes of most cells, causing neurodegeneration and dysfunction of lung and liver. 154
Lysosomes are also key in lipid hydrolysis ensured by autophagy, i.e., “lipophagy”. 155 Activity of lysosomal acid lipases involved in lipophagy requires low pH. 156 In hepatocytes, lipophagy contributes to the degradation of intracellular LDs and to the release of FFA. In line with this, patients with TMEM199 and CCDC115 mutations present hypercholesterolemia and fatty liver disease. 157 Impaired lipophagy also seems involved in VMA21-CDG: hepatocytes from patients show intracellular accumulation of LDs, which can be found in autolysosomes or lysosome-like structures. 8 LD accumulation was also reported in XMEA fibroblasts, although LD-containing autolysosomes have never been described. 8 Fibroblasts from VMA21-CDG patients and, to a lesser extent, from XMEA patients, also exhibited a build-up of cholesterol. 8 Bafilomycin A treatment exacerbated LD and cholesterol accumulation in control and XMEA fibroblasts. In contrast, bafilomycin A did not further increase LD and cholesterol accumulation in VMA21-CDG hepatocytes. 8 This suggested different pathomechanisms depending on the mutation in VMA21 and/or the cell type. Of note, cholesterol retention in lysosomes reduced its amount in the ER, secondarily promoting the lipogenic pathway: levels of sterol response element-binding protein-1 (SREBP1), a sensor of cholesterol in ER, increased in VMA21-CDG but not in XMEA fibroblasts. Upregulation of SREBP1-mediated lipogenesis may hence contribute to the elevated levels of plasma cholesterol in VMA21-CDG patients. 8
ER stress
ER stress arises from an accumulation of unfolded or misfolded proteins in the ER, which usually results from an imbalance between protein synthesis, maturation and intracellular trafficking. 158 The pathogenic role of ER stress has been established in several disorders, such as neurodegenerative diseases, diabetes and liver cirrhosis.159–161 ER stress, in particular in muscle fibres, can trigger autophagy induction, which may then contribute to the elimination of unfolded proteins.162–164 However, ER stress also reduced lysosomal function in muscle fibres, 164 suggesting that it may rather limit autophagic flux. Of note, reduced levels of VMA21 were detected upon ER stress induction in muscle fibres. 164
In CDG fibroblasts, VMA21 deficiency resulted in activation of the unfolded protein response (UPR) associated with protein kinase R-like ER kinase (PERK), suggesting ongoing ER stress. 8 Deficiency for the chaperone ATP6AP2 in Drosophila also led to ER stress, which may arise from the defective assembly of the v-ATPase V0 domain and its potential degradation within the ER.165,166 Whether VMA21 chaperones the assembly and/or folding of other proteins or complexes in the ER remains unknown. In this context, VMA21 deficiency may induce ER stress by dysregulating the maturation of several proteins, including v-ATPase V0 domain. Although ER stress and UPR activation were not detected in fibroblasts from one XMEA patient analyzed by Cannata et al., 8 they remain to be investigated in muscle cells from XMEA patients.
Conclusion
Interest to VMA21 has grown these last years with the expansion of the clinical spectrum associated with VMA21 mutations. The reported role of VMA21 in the assembly of the v-ATPase pump is consistent with pathogenic involvement of autophagy, glycosylation, lipid metabolism, and/or ER stress in XMEA and VMA21-CDG. The comparison between XMEA and VMA21-CDG uncovered differences in the clinical features and pathomechanisms of the two diseases. While ER stress and glycosylation defects were not observed in XMEA patients, impaired autophagy and increased cholesterol were detected in both VMA21-CDG and XMEA cells. Residual levels of VMA21, and whether one or the two VMA21 isoforms are affected by the mutation, may determine the processes impaired and/or the tissue clinically targeted. Hence, further investigations are required to clarify the respective contribution of VMA21 isoforms in the pathogenesis of XMEA and VMA21-CDG, and the pathomechanisms underlying the clinical differences between the two diseases. In line with this, systematic evaluation of the expression of VMA21-120 (in addition with VMA21-101) in patients with CDG or XMEA, as well as the identification of other targets/partners of each isoform, are crucial. The role of circVMA21 in XMEA and VMA21-CDG should also be considered in this puzzle. Future research development on these mechanistic aspects will ultimately help designing common or specific therapeutic interventions for VMA21-related diseases.
Footnotes
Abbreviations
Funding
P.C. was financed with a grant from the Swiss National Science Foundation (PCEFP3_181102).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.