
Abstract
Background
In recent years, treatments have been approved for certain neuromuscular diseases. In some cases, early pre-symptomatic treatment is necessary for optimal response, and thus newborn screening is critical.
Objective
To review the current status of newborn screening programs for neuromuscular diseases and early diagnosis through genetic testing.
Methods
Following the PRISMA guidelines, a literature search was performed on PubMed for screening of neuromuscular diseases; the search was conducted on literature available as of 1 May 2024.
Results
Included were 77 articles on newborn screening for seven diseases: spinal muscular atrophy (19 studies), Duchenne muscular dystrophy (15), Pompe disease (20), X-linked adrenoleukodystrophy (14), Krabbe disease (6), metachromatic leukodystrophy (2), and myotonic dystrophy 1 (1). Ten articles on rapid genomic diagnosis were identified.
Conclusion
Since 2021, newborn screening programs for neuromuscular diseases have been established, notably in X-linked adrenoleukodystrophy, spinal muscular atrophy, Pompe disease, and Duchenne Muscular Dystrophy. Even in diseases where treatment is currently not life-changing, such as Krabbe disease, new newborn screening programs continue to be implemented, especially in the USA. The use of genetic diagnostic tests does not yet appear to be widespread or at least not widely reported. As new treatments become available, genomic newborn screening programs will need to be rapidly and broadly implemented.
Keywords
Introduction
The treatment of patients with neuromuscular diseases (NMD) has evolved considerably in recent years as disease-modifying treatments have been approved for clinical use for patients with several of the approximately 400 described NMDs. The diagnosis of an NMD is frequently delayed due to the rarity and complexity of these diseases.1–4 This delay can result in irreversible muscle damage that can greatly reduce the effectiveness of treatment when available.5–7 Even in cases where there is no muscle destruction, such as certain forms of congenital myasthenia, the lengthy diagnostic process can lead to decades of compromised quality of life before a correct diagnosis is made and suitable treatment is initiated as well as secondary complications like scoliosis or chest infection. 8
Improvements of the standard of care and diagnosis workflow, establishment of neuromuscular centers, and increased availability of genomic methods for testing have shortened the diagnostic odyssey. 9 Diagnostic delays remain a real issue, however, and are especially harmful for patients who suffer from treatable conditions. Newborn screening (NBS) is a very efficient way of identifying treatable diseases. NBS has been in use for nearly 60 years for various diseases. Incorporation of a test within an NBS program is based on a set of criteria proposed by Wilson and Jungner, 10 with interpretations varying from country to country, particularly as regards to the criterion that a treatment be available. In Europe, for example, the approach is conservative with only diseases that are treatable included, whereas in the United States of America (USA) a more flexible approach has been adopted. 11
NBS panels previously included only biochemical or metabolic analyses and therefore could not include tests for diseases without metabolic or endocrine markers such as NMDs. Driven by the availability of effective treatment for certain NMDs, genetic NBS is developing very rapidly. A notable example is spinal muscular atrophy (SMA). In just a few years use of a genetic NBS test has become common in high-income countries. The aim of this scoping review is to describe the current status of NBS and early diagnosis of NMD. We carried out a scoping review over the last 10 years to assess the contribution of genetic testing by NBS or whole genome sequencing (WGS) for NMD.
Methods
Literature search
We followed the PRISMA checklist 12 and conducted a thorough literature search using Medline (PubMed). We sought to identify original, full-text articles that discussed NBS tests for NMDs that were published after 1 January 2014 and before 1 May 2024. To identify these articles, we used key terms related to NBS such as ‘neonatal screening’, ‘dried blood’, and ‘Guthrie’, and those related to early diagnosis such as ‘early diagnosis’, ‘whole genome sequencing’ in combination with relevant neuromuscular disease keywords. A detailed search strategy is provided in Supplementary file 1.
Selection of studies
We used the Covidence application to screen the articles. To be included, articles had to be full articles, in English or French, published after 1 January 2014 that had been peer reviewed. Articles had to describe established NBS programs or pilot projects for NMD or describe diagnoses made within the first 28 days of a child's life. Three reviewers (TD, HL, NB) first screened titles and abstracts for eligibility with the consensus of a minimum of two reviewers required for inclusion. Then two reviewers (TD, HL) reviewed the full text. Reasons for exclusions were recorded. Any potential disagreements were resolved by consensus. When more than one article described the same pilot project, only the most recent was included. We retained studies demonstrating the efficacy of NBS on deidentified Guthrie cards only if there were no pilot projects with identified patients.
Data extraction and presentation
Data were extracted using the Covidence application. Articles were then classified into two categories: NBS or early diagnosis in symptomatic patients. Articles were then sorted by disease.
Results
Study selection process
The initial searches identified 772 articles describing NBS for neuromuscular disorders. They were screened by title and abstract, and 183 articles were identified for full-text screening. Full-text screening identified 105 articles; 21 articles were added based on reviews of bibliographies or after additional searches of more recent publications. Of these 39 articles were excluded because they described the same studies. In total 77 articles on screening for NMD and 10 on early diagnosis were included in this review. Supplementary file 2 shows the flow chart based on the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines used for the identification of these studies.
Newborn screening programs for NMDs
Spinal muscular atrophy
SMA is a recessive disease caused by homozygous loss-of-function mutations (usually deletions of exon 7) in SMN1, the gene encoding a protein critical for maintenance of motor neurons. Three drugs are approved for treatment of SMA: nusinersen, 13 onasemnogene-abeparvovec,14,15 and risdiplam. 16 Extensive data demonstrated that these drugs are most efficacious if treatment begins before symptom onset.6,17
NBS for SMA was reported in 19 articles, which describe 19 studies each conducted in a region or country.18–36 The initial implementation of pilot NBS programs for SMA took place in Taiwan in 2014, 37 followed by New York in 2016. 38 Interestingly, these pilot programs were conducted before any drugs were approved for use in these patients, and some of the patients identified in these pilot programs were included in clinical trials.36,39 The incidence rates vary significantly: For example, in Italy, a rate of 1 in 6000 was reported, 40 whereas in Ontario the reported rate was 1 in 28,000. 41 One of the lowest incidence rates of the disease was found in New York, 42 which may be due to increased use of preconception screening due to increased communication about the disease. 43 In most programs, the first tier of screening is done by quantitative real-time polymerase chain reaction (qPCR), and the second and confirmatory tests are by multiplex ligation-dependent probe amplification (MLPA) or digital droplet PCR. Some programs had issues with false positives, especially at the beginning of the program, and the reasons have been identified and corrected. For example, heparin was used in the tubes used for sampling. 28 In recent years, there has been exponential growth in the use of NBS for SMA, and most high-income countries now screen newborns for this disease. 44 A recent survey showed that 100% of newborns in the USA and 66% of newborns in Europe now screened for SMA. 45 Table 1 summarizes the articles that describe NBS for SMA.
NBS for SMA.
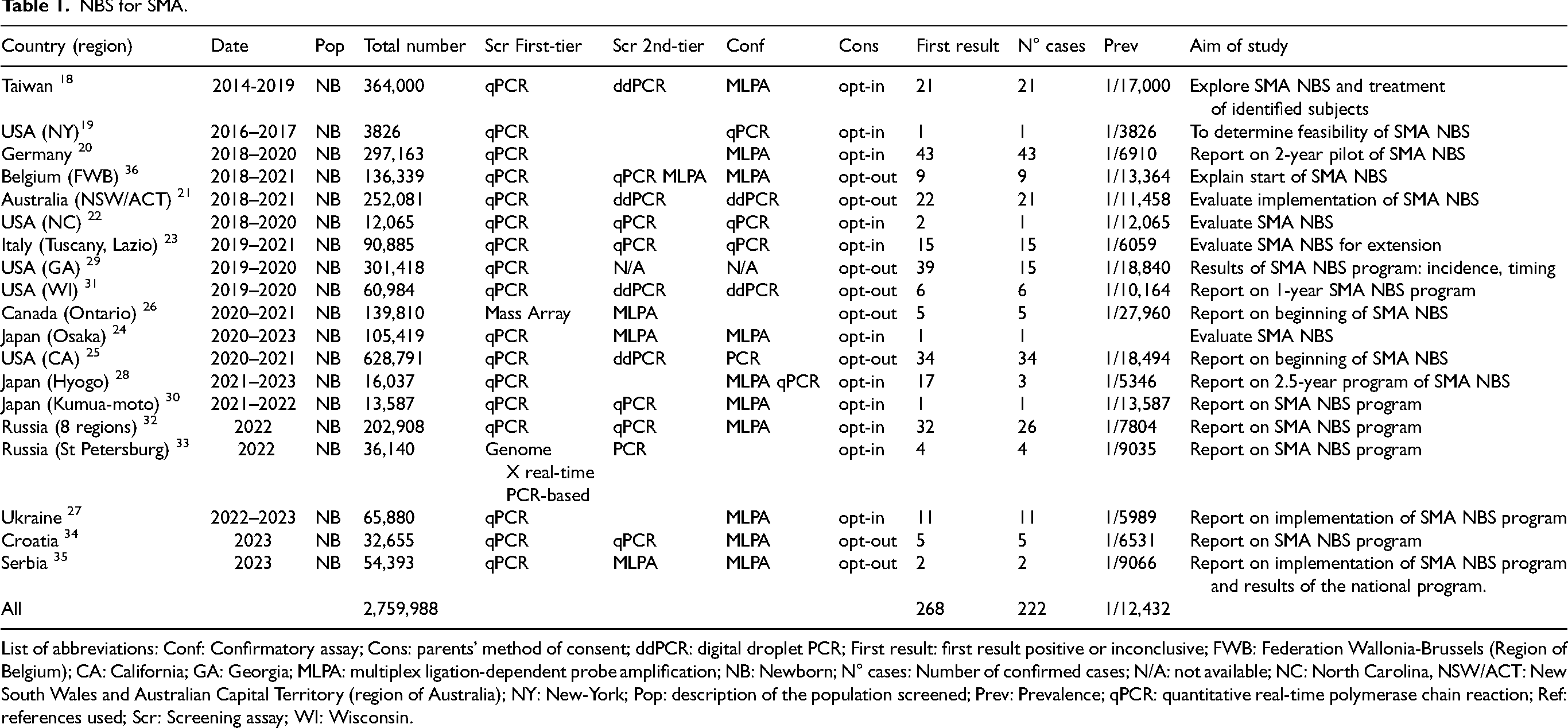
List of abbreviations: Conf: Confirmatory assay; Cons: parents’ method of consent; ddPCR: digital droplet PCR; First result: first result positive or inconclusive; FWB: Federation Wallonia-Brussels (Region of Belgium); CA: California; GA: Georgia; MLPA: multiplex ligation-dependent probe amplification; NB: Newborn; N° cases: Number of confirmed cases; N/A: not available; NC: North Carolina, NSW/ACT: New South Wales and Australian Capital Territory (region of Australia); NY: New-York; Pop: description of the population screened; Prev: Prevalence; qPCR: quantitative real-time polymerase chain reaction; Ref: references used; Scr: Screening assay; WI: Wisconsin.
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is the most prevalent childhood form of muscular dystrophy. This condition arises from deletions, duplications, or single nucleotide variants that affect the production of functional dystrophin. First symptoms typically occur when children are 2–3 years old and include proximal weakness and muscle pseudo-hypertrophy. The weakness progresses rapidly and results in the loss of walking between the ages of 7 and 15. On average, there is a two-year gap between the onset of symptoms and diagnosis of DMD. 1 This diagnostic delay not only causes distress for parents, but it can also result in a delay in appropriate care like steroids and multidisciplinary follow-up.
The Food and Drug Administration (FDA) has approved eight drugs for use in patients with DMD: two steroids (deflazacort and vamorolone), four oligo antisense nucleotide that aim to skip exons 51 (eteplirsen, vitolarsen), 53 (golodirsen) and 45 (casimersen), a gene therapy that provides a copy of microdystrophin using an AAV-microdystrophin construct, and the histone deacetylase inhibitor givinostat. There are age restrictions on some of these drugs. For example, givinostat is for use in patients 6 years and older and the gene therapy is for use in patients 4 years and older. The exon skipping drugs do not have age restrictions. Clinical and pre-clinical efforts toward development of a number of additional potential therapies are ongoing. 46
Our literature search identified 13 pilot projects or official implementations of NBS for DMD between 1974 and 2022 described in 15 articles.47–60 New pilot programs were recently launched in the USA; the main objectives are to evaluate the feasibility and benefit of NBS for DMD.51,61 In the USA, there is considerable pressure from parent associations to add DMD to the list of diseases in the Recommended Uniform Screening Panel (RUSP), even though evidence for efficacies of available treatments is limited. One of the rational of this position is to avoid recurrence of the disease in the family. In screening efforts reported to date, the incidence in young males is between 1 in 3500 48 to 1 in 10,300. 49 Table 2 summarizes the articles that describe NBS for DMD.
NBS for DMD.
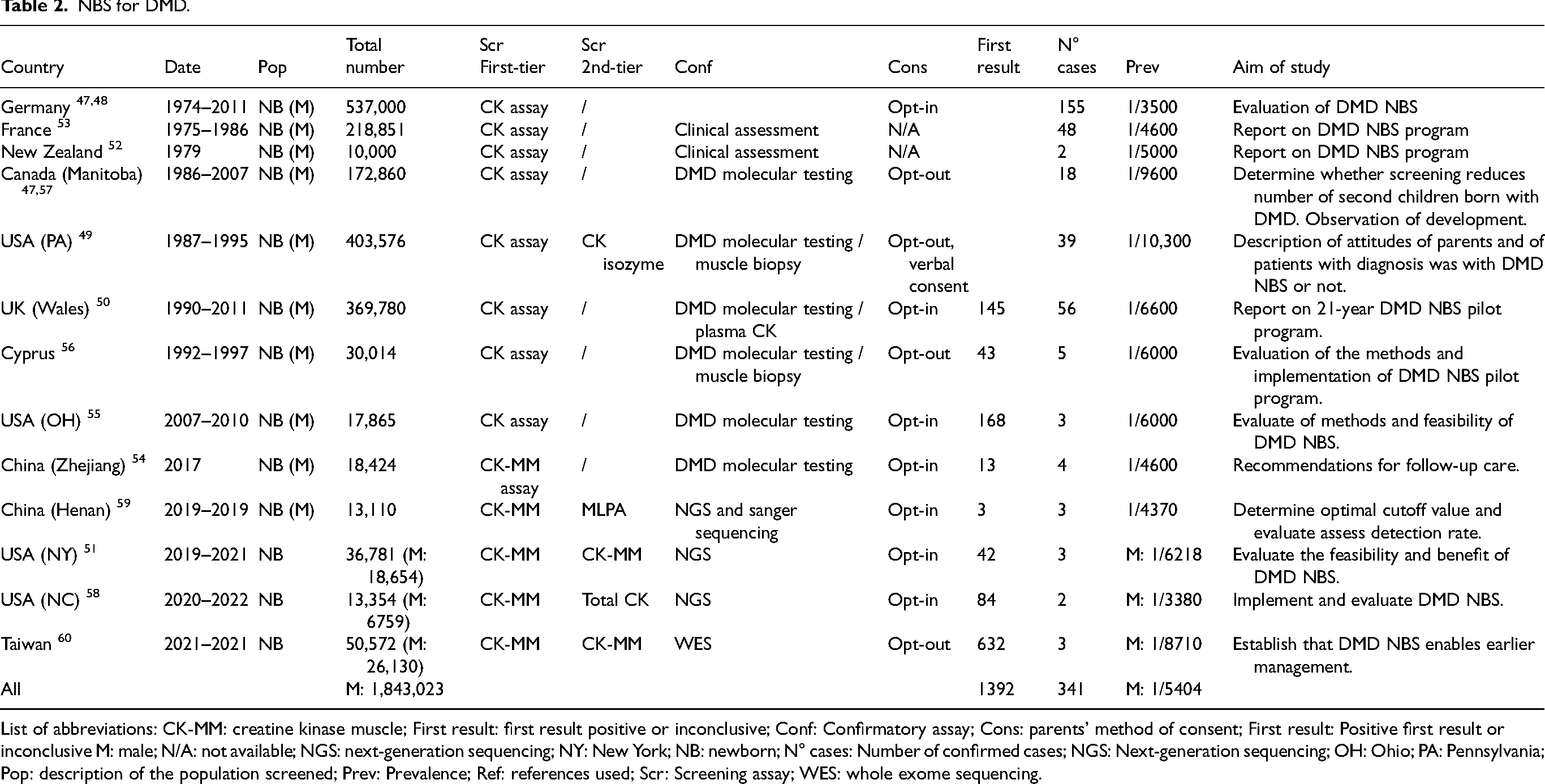
List of abbreviations: CK-MM: creatine kinase muscle; First result: first result positive or inconclusive; Conf: Confirmatory assay; Cons: parents’ method of consent; First result: Positive first result or inconclusive M: male; N/A: not available; NGS: next-generation sequencing; NY: New York; NB: newborn; N° cases: Number of confirmed cases; NGS: Next-generation sequencing; OH: Ohio; PA: Pennsylvania; Pop: description of the population screened; Prev: Prevalence; Ref: references used; Scr: Screening assay; WES: whole exome sequencing.
X-linked adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal impairment caused by a loss of function mutation in the ABCD1 gene located on the X chromosome. This metabolic disorder affects the adrenal glands, brain, and spinal cord, resulting in devastating consequences. Boys who are hemizygous are more severely affected by X-ALD than are heterozygous girls (60% of cases). If left untreated, subjects with X-ALD typically survive only a few years after symptom onset. Treatments include corticosteroids for adrenal insufficiency, hematopoietic stem cell transplantation, and gene therapy. Treatment during the initial stages of brain inflammation prior to neurologically debilitating brain adrenoleukodystrophy enhances survival rates and improves functional outcomes. 62
NBS for X-ALD was reported in 14 articles that describe 13 studies, each conducted in a different region or country,63–67 most of them in the USA.62,68–75 Most of these articles were published within the last five years, a sign of the rapid development of NBS programs of X-ALD. In the USA, the program, which was first implemented in 2016, has proven to be effective. The first-tier tests for X-ALD are all metabolic, but confirmatory tests are increasingly genetic. The incidence of X-ALD in males varies from 1 in 4000 76 to 1 in 14,000. 69 Table 3 summarizes the articles that describe NBS for X-ALD.
NBS for X-ALD.
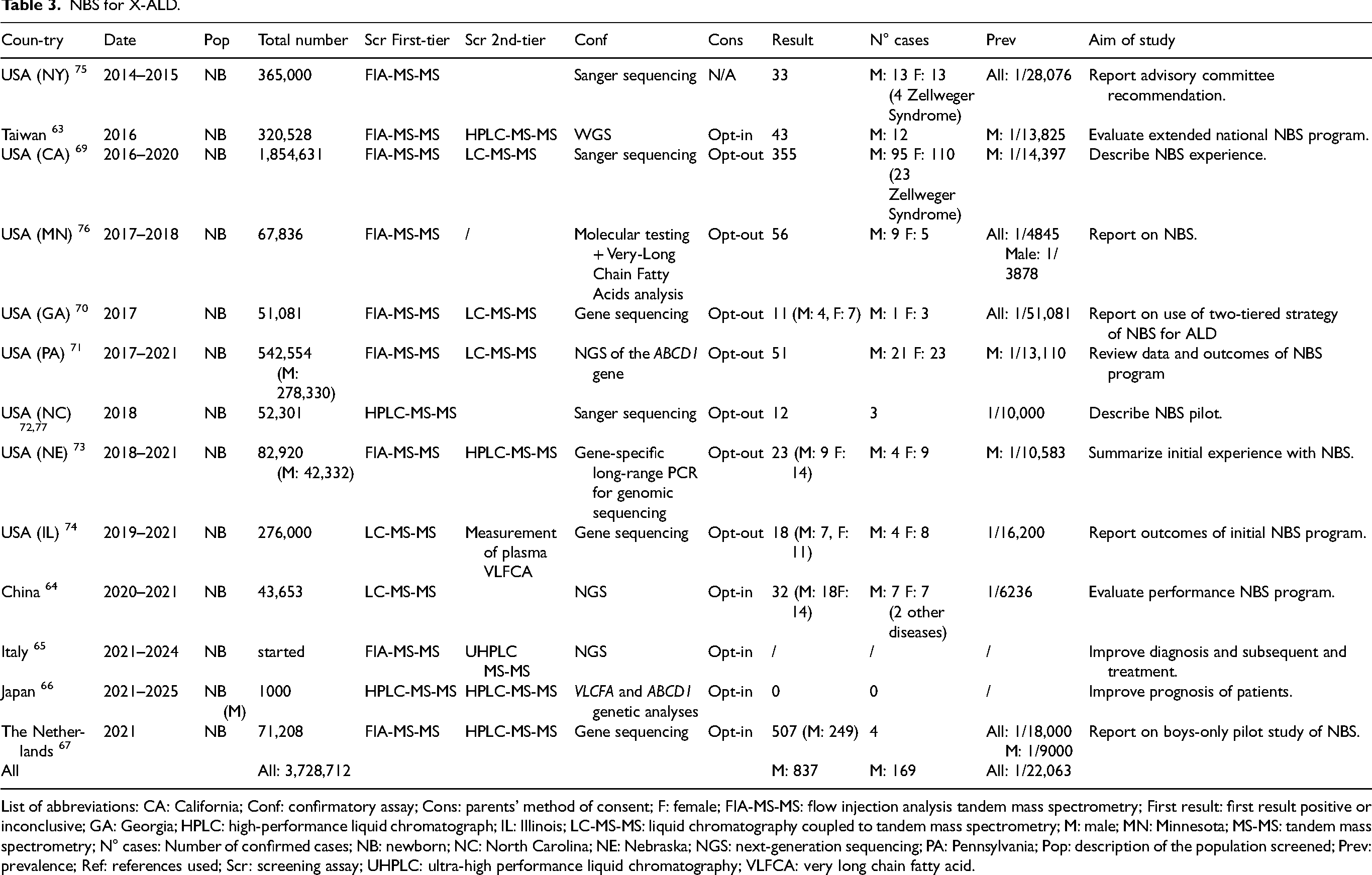
List of abbreviations: CA: California; Conf: confirmatory assay; Cons: parents’ method of consent; F: female; FIA-MS-MS: flow injection analysis tandem mass spectrometry; First result: first result positive or inconclusive; GA: Georgia; HPLC: high-performance liquid chromatograph; IL: Illinois; LC-MS-MS: liquid chromatography coupled to tandem mass spectrometry; M: male; MN: Minnesota; MS-MS: tandem mass spectrometry; N° cases: Number of confirmed cases; NB: newborn; NC: North Carolina; NE: Nebraska; NGS: next-generation sequencing; PA: Pennsylvania; Pop: description of the population screened; Prev: prevalence; Ref: references used; Scr: screening assay; UHPLC: ultra-high performance liquid chromatography; VLFCA: very long chain fatty acid.
Pompe disease
Pompe disease, also called glycogenosis type 2, is an autosomal recessive lysosomal storage disorder resulting from acid alpha-glucosidase (GAA) deficiency. The disease has a broad clinical spectrum, ranging from the infantile form, which manifests within the first few months of life, to adult forms. Without treatment, the infantile form invariably results in early mortality due to cardiorespiratory failure or respiratory infection, typically before the age of one year. In the later-onset forms, symptoms can emerge at any age and are linked to progressive skeletal muscle dysfunction.
Enzyme replacement therapy (ERT) with alglucosidase alfa (Myozyme) received approval from the FDA and the European Medicines Agency (EMA) in 2006. 78 In 2023, the FDA approved the combination therapy of cipaglucosidase alfa-atga with miglustat to treat adults who are not improving on their current regime of ERT. 79 Early intervention with ERT is associated with improved outcomes. 80 Gene therapy development is mostly focused on restoring GAA production and is currently in clinical development.
NBS for Pompe disease was reported in 20 articles describing 18 studies. The inaugural NBS pilot program was launched in Taiwan in 2005 and has been consistently documented since.81,82 Since 2015, Pompe disease has been included in the RUSP in the USA and screening for Pompe disease is part of NBS programs in 38 states. 83 These efforts have been described in a number of publications.84–92 Comparable initiatives have been established in Italy, 93 Mexico, 94 Japan,95,96 China, 97 and Brazil.98–100 The reported incidences for infantile and late forms vary from 1:10,600 in Brazil to 1:38,000 in Japan. Table 4 summarizes the articles that describe NBS for Pompe disease.
NBS for pompe disease.
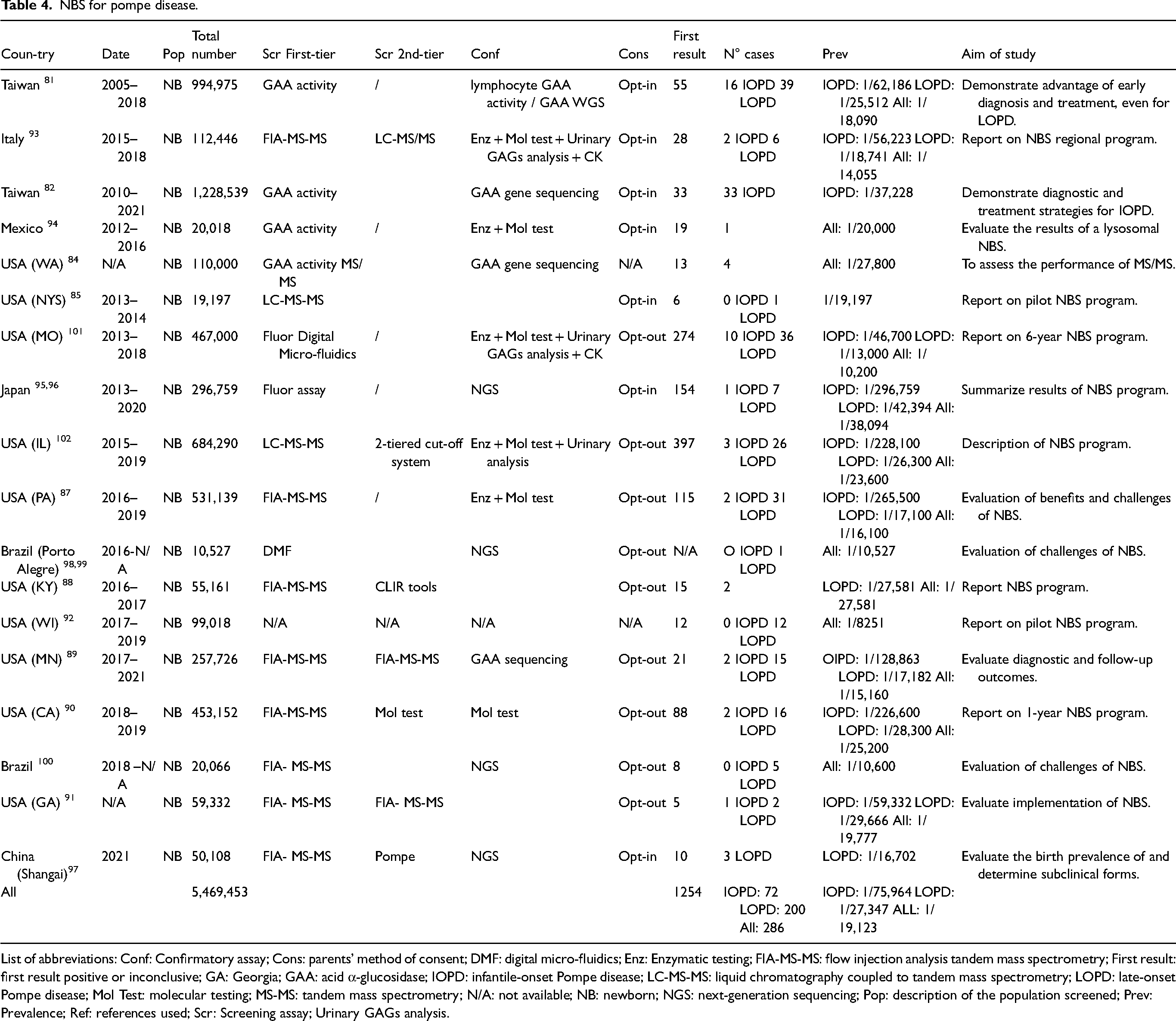
List of abbreviations: Conf: Confirmatory assay; Cons: parents’ method of consent; DMF: digital micro-fluidics; Enz: Enzymatic testing; FIA-MS-MS: flow injection analysis tandem mass spectrometry; First result: first result positive or inconclusive; GA: Georgia; GAA: acid α-glucosidase; IOPD: infantile-onset Pompe disease; LC-MS-MS: liquid chromatography coupled to tandem mass spectrometry; LOPD: late-onset Pompe disease; Mol Test: molecular testing; MS-MS: tandem mass spectrometry; N/A: not available; NB: newborn; NGS: next-generation sequencing; Pop: description of the population screened; Prev: Prevalence; Ref: references used; Scr: Screening assay; Urinary GAGs analysis.
Krabbe disease
Krabbe disease is an autosomal recessive lysosomal disorder that impacts the white matter of both the central and peripheral nervous systems. The severity depends on the age of onset. This disease is caused by loss-of-function mutations in both alleles of the GALC gene, leading to a deficiency in galactosylceramidase. Historically, 85–90% of patients are diagnosed with the infantile form, the most severe type, which manifests within the first six years of life. In cases where the disease begins within the first year, rapid neurological decline is observed, often resulting in death before the age of two. Late-onset Krabbe disease presents with much more variability in symptoms and progression. 103 The rarity of the disease poses a challenge for evaluating the effectiveness of pre-symptomatic treatments. Krabbe disease has proven resistant to most single-therapy treatments. Hematopoietic stem cell transplantation, the current standard of care, can modify the disease's progression, but it only slows it down, even if begun pre-symptomatically. Recent successes with combination therapies in small animal models and the discovery of new pathogenic mechanisms offer hope for developing effective treatments for this devastating condition.104,105
Six studies have evaluated NBS for Krabbe disease: three in the USA,88,106,107 one in Mexico, 94 one in China, 97 and one in Brazil. 100 Given the low incidence of the disease, estimated at 1 in 100,000, 105 not all programs have identified cases. Attempts to include Krabbe on the RUSP have been twice unsuccessful and in January of 2024, the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) officially recommended that Krabbe disease be added to the RUSP. It is currently implemented in only eleven states in the USA. Recent publications have raised ethical concerns regarding such screening, 108 even though patient associations are pushing for its implementation. 109 Table 5 summarizes the articles that describe NBS for Krabbe disease.
NBS for Krabbe disease, MLD, and MD1.
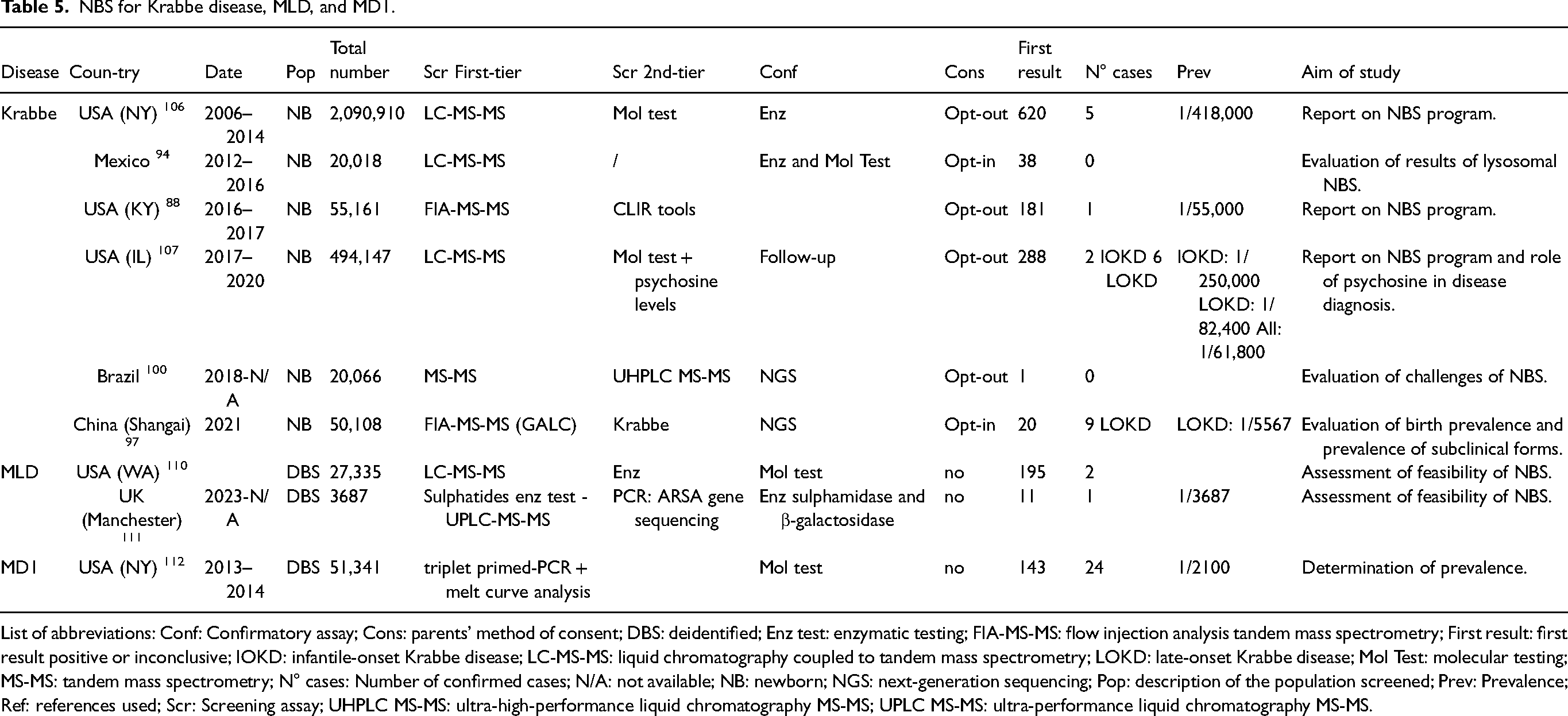
List of abbreviations: Conf: Confirmatory assay; Cons: parents’ method of consent; DBS: deidentified; Enz test: enzymatic testing; FIA-MS-MS: flow injection analysis tandem mass spectrometry; First result: first result positive or inconclusive; IOKD: infantile-onset Krabbe disease; LC-MS-MS: liquid chromatography coupled to tandem mass spectrometry; LOKD: late-onset Krabbe disease; Mol Test: molecular testing; MS-MS: tandem mass spectrometry; N° cases: Number of confirmed cases; N/A: not available; NB: newborn; NGS: next-generation sequencing; Pop: description of the population screened; Prev: Prevalence; Ref: references used; Scr: Screening assay; UHPLC MS-MS: ultra-high-performance liquid chromatography MS-MS; UPLC MS-MS: ultra-performance liquid chromatography MS-MS.
Metachromatic leukodystrophy
Metachromatic leukodystrophy (MLD) is a metabolic disorder inherited in an autosomal recessive manner, characterized by the accumulation of sulfatides in the central and peripheral nervous systems due to a deficiency in the enzyme arylsulfatase A, resulting in demyelination. The disease primarily manifests as a decline in motor or cognitive functions. Depending on the subtype, the disease has a highly variable progression and duration but eventually leads to severe disability and death. Both the EMA and the FDA have approved atidarsagene autotemcel, an autologous hematopoietic stem cell gene therapy, for the treatment of children diagnosed with pre-symptomatic late infantile, pre-symptomatic early juvenile, or early symptomatic early juvenile MLD.
The first pilot project aimed at evaluating the feasibility of NBS for MLD was conducted in the USA in 2020, screening 27,335 de-identified dried blood spots. 110 Very recently, a study in Manchester, UK showed the effectiveness of a sulphate test on dried blood spots and the evaluation of ARSA enzyme activity as a two-tier strategy. 111 One infant was identified. This program could soon be recommended by the UK National Screening Committee for incorporation into routine NBS. Table 5 summarizes the articles that describe NBS programs for MLD.
Myotonic dystrophy 1
Myotonic dystrophy 1 (MD1) is an autosomal dominant disorder marked by muscle weakness, myotonia, early onset cataracts, and various systemic symptoms affecting the brain, endocrine system, heart, gastrointestinal tract, uterus, skin, and immune system that differ based on age of onset. The disorder is caused by a pathological expansion of over 50 CTG repeats in the DMPK gene. MD1 onset occurs earlier with each successive generation, which typically happens with maternal transmission. The most severe form, congenital MD1, occurs in 15% of cases. Patients present with severe generalized weakness at birth, respiratory distress, hypotonia, and feeding difficulties. Affected infants may experience delayed cognitive and motor development, intellectual disability, and autism spectrum disorder, with the physical symptoms potentially leading to fatal outcomes. The incidence ranges from 0.5 to 1.8 per 100,000.
Oligonucleotide-based agents and gene therapies have been tested in pre-clinical models and are currently in clinical development. Nevertheless, no specific disease-modifying treatments are currently approved. Management primarily involves monitoring for complications and providing assistive devices, hormone therapy, and pain management.
We identified an article describing a pilot project in the USA that utilized deidentified dried blood spots to determine the prevalence of MD1. The study found a prevalence rate of 1 in 21,100. 112 Table 5 summarizes this article.
Rapid genomic diagnosis
Exome and genome sequencing is increasingly used in neonatal intensive care units to diagnose and treat critically ill infants, but the gene panels used for these purposes are poorly reported and therefore little is known regarding the efficacy of this type of testing. The conventional diagnostic approach based on hypotheses might not be effective in these situations, as clinical manifestations during the neonatal period are often nonspecific. Moreover, phenotypes linked to reduced survival in numerous genetic disorders may go unrecognized due to early mortality. In subjects with NMDs, exome and genome sequencing can enable diagnosis to be made. We identified articles reporting two pilot projects in the USA,113,114 two in India,115,116 two in China,117,118 one in South Korea, 119 one in Sweden, 120 one in the United Emirates Arab, 121 and one in Belgium 122 that evaluated use of genetic testing for diagnosis of NMDs.
Five teams have reported unique cases diagnosed thanks to genetic testing.113,115,116,120,121 These diagnoses enabled either early treatment, as was the case for patients with myopathy in Shwachman-Diamond syndrome, a neonatal-onset MYH2 hereditary myosin myopathy, and congenital hypomyelinating neuropathy, or explained early death (as was the case for a patient with a severe perinatally lethal neuromuscular form of glycogen storage disease IV). 116 Five teams have targeted specific populations: hypotonic children, children of low birth weight compared with gestational age, or children with seizures, recruited from neonatal intensive care units, paediatric intensive care units and paediatric neurology units.114,117–119,122 In these studies, genetic testing used targeted panels or more extensive whole exome or whole genome sequencing with the goal of evaluating diagnostic speed and accuracy. In the specific targeted populations, the diagnoses yield was between 27 and 63%, with peripheral hypotonia disorders, Krabbe disease, cerebrotendinous xanthomatosis, SMA, or congenital myasthenic syndrome identified Table 6.
Rapid genomic diagnosis of NMD.
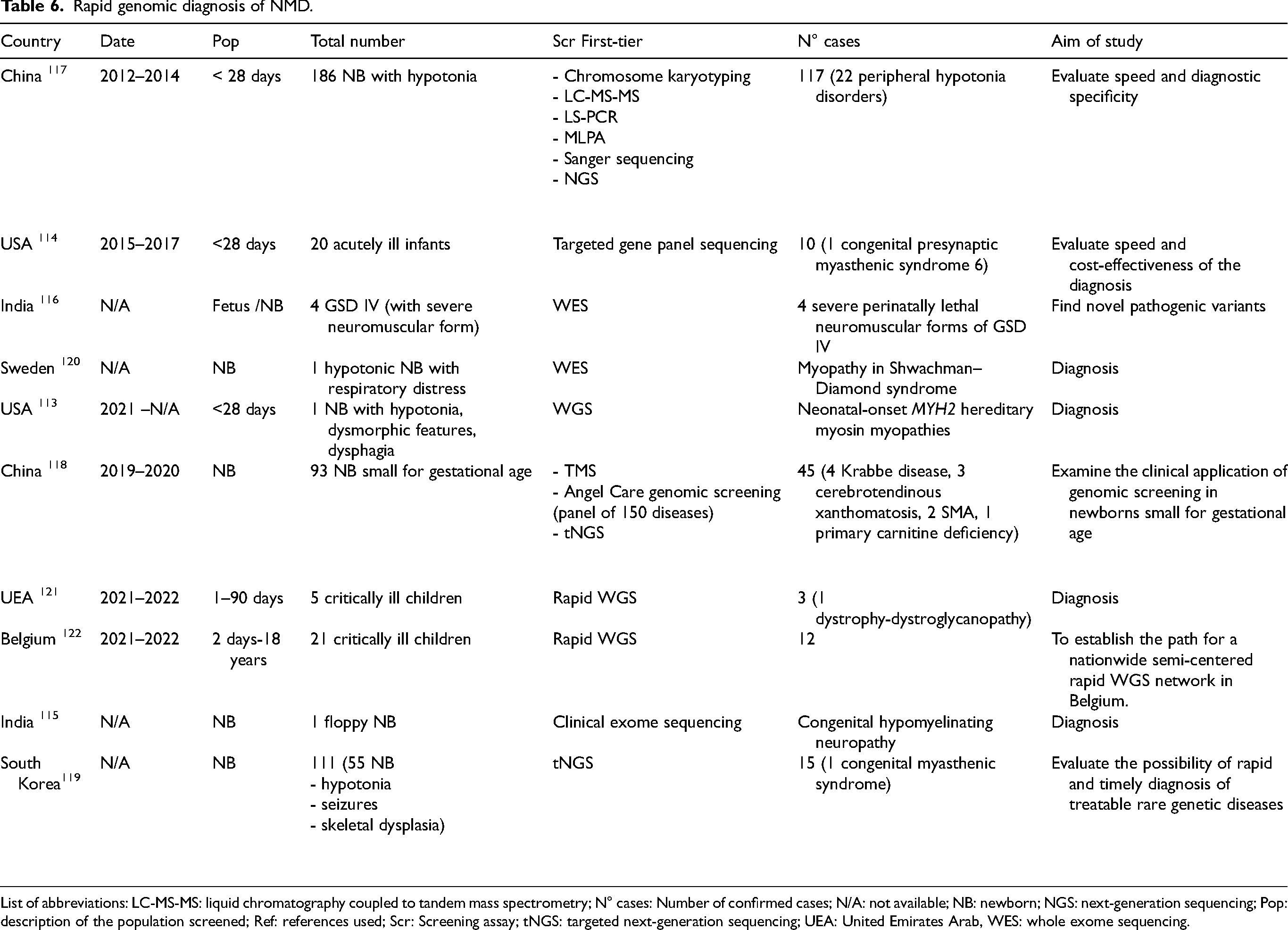
List of abbreviations: LC-MS-MS: liquid chromatography coupled to tandem mass spectrometry; N° cases: Number of confirmed cases; N/A: not available; NB: newborn; NGS: next-generation sequencing; Pop: description of the population screened; Ref: references used; Scr: Screening assay; tNGS: targeted next-generation sequencing; UEA: United Emirates Arab, WES: whole exome sequencing.
Discussion
Our literature search identified 87 articles describing 82 early identification programs for neuromuscular diseases: 72 studies evaluated NBS programs for seven NMDs and ten studies focused on early genetic diagnosis for various diseases including NMDs. The fast-growing number of peer-reviewed papers in this field illustrates the growing understanding of the importance of early, and ideally pre-symptomatic, identification of patients with treatable conditions such as X-ALD, SMA, Pompe disease, and DMD. These diseases can be diagnosed with a simple biochemical test or straightforward genetic test. Congenital myasthenic syndrome is more difficult to diagnose, and we anticipate similar issues in limb-girdle muscular dystrophies for which treatments are in development but no simple and specific tests are available.
When there are no sensitive biochemical assays nor specific hotspot mutations (such as the deletion of exon 7 of SMN1 in patients with SMA), which is the case for the majority of NMDs, screening for these disorders will not be compatible with the biochemical methods used in current NBS programs. The fast-paced development of therapies also necessitates the ability to quickly add a gene to a genomic NBS program to ensure that patients are diagnosed as rapidly as possible. Some are hesitant about genomic NBS for several reasons. One primary concern is the potential for false positives or uncertain results, which can cause unnecessary stress and anxiety for families. Additionally, the interpretation of genetic data is complex, and not all detected variants are fully understood, leading to potential misdiagnoses or overdiagnoses. Enabling quicker diagnoses allows for more timely treatments, preventing long-term complications and significantly improving the effectiveness of interventions. For these reasons, several genomic NBS programs are currently underway that use either using WGS or targeted next-generation sequencing. 123 Although the panels differ, all screen for mutations in genes implicated in NMDs such as MLD. Parents appear positive about these first genomic NBS programs. 124 As the cost of adding a new disease is not a limitation when WGS is employed, we expect that many NMDs will be added to the list of diseases evaluated in NBS programs over the next few years. As both genomic NBS and early diagnostic testing will eventually utilize WGS, it is foreseeable that raw data of WGS performed for NBS could be in the future re-analysed for diagnosis in a child presenting any serious condition.
This scoping review has certain limitations. First, only one database (Medline) was consulted. We also restricted our search to original articles; conference presentations were not considered. Data presented at neuromuscular disease conferences are likely to be published quickly, however, as the neuromuscular world is undergoing a period of transition due to the recent approval of effective treatments.
Conclusion
Since 2021, newborn screening for NMDs, notably in X-ALD, SMA, and Pompe disease, have been incorporated in routine NBS programs. Even for diseases where treatment is currently not life-changing, such as Krabbe disease, new NBS programs continue to be implemented, especially in the USA. The use of genetic diagnostic tests does not yet appear to be widespread, or at least not widely reported. The genomic newborn screening programs currently underway will undoubtedly be a lever for rapid development. Rapid incorporation of tests into routine NBS will be critical as new treatments for NMDs become available.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602241296286 - Supplemental material for Newborn screening and rapid genomic diagnosis of neuromuscular diseases
Supplemental material, sj-docx-1-jnd-10.1177_22143602241296286 for Newborn screening and rapid genomic diagnosis of neuromuscular diseases by Tamara Dangouloff, Helena Lang, Noor Benmhammed and Laurent Servais in Journal of Neuromuscular Diseases
Supplemental Material
sj-docx-2-jnd-10.1177_22143602241296286 - Supplemental material for Newborn screening and rapid genomic diagnosis of neuromuscular diseases
Supplemental material, sj-docx-2-jnd-10.1177_22143602241296286 for Newborn screening and rapid genomic diagnosis of neuromuscular diseases by Tamara Dangouloff, Helena Lang, Noor Benmhammed and Laurent Servais in Journal of Neuromuscular Diseases
Footnotes
Abbreviation
Acknowledgments
The authors thank Jacky Wyatt for language editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HL and NB have no conflicts of interest to report. TD has given lectures sponsored by Biogen and Roche. LS Servais has given consultancy - attended the board of Zentech and Illumina. Outside the scope of the paper, LS has given consultancy - attended the board for Roche, Biogen, Novartis, Scholar Rock, BioHaven, Pfizer, PTC, Dyne, Wave, Biomarin, Myostana, MetrioPharma, Astellas, Sanofi, Sysnav, and RegenexBio.
Data availability
The data supporting the findings of this study are available within the article and/or its supplementary material.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.