
Abstract
Highly efficacious, potentially curative gene therapies holds immense clinical promise, but also present complex challenges. At the time of regulatory approval and health technology assessment (HTA), evidence of efficacy and safety of gene therapies is often uncertain. In addition, research, development, and manufacturing costs, small pools of eligible patients, and the fact that many gene therapies are administered only once means that they frequently are associated with very high “one-off” price points. Although only a limited number of products have been brought to market globally, hundreds of clinical trials of gene therapies, including several of monogenetic neuromuscular diseases, are currently ongoing. Over time, as more and more conditions become amendable to gene therapy, the number of transformative, high-cost treatments is likely to increase considerably. For these reasons, concerns have been raised regarding the suitability of current health policy systems, including HTA frameworks, in ensuring appropriate access to these therapeutic innovations while simultaneously safeguarding value for taxpayers’ money, as well as affordability and sustainability. This review provides a summary overview of current challenges and future perspectives of gene therapies for neuromuscular diseases from a health economic point of view.
Keywords
Introduction
Gene therapy, defined as the “the intentional, expected permanent, and specific alteration of the DNA sequence of the cellular genome, for a clinical purpose” [1], marks a paradigm shift in the treatment of disease in humans. Indeed, the emergence of durable and potentially curative gene therapies has dramatically altered the prognosis of several severely debilitating and ultimately fatal illnesses. Monogenetic neuromuscular diseases have been in the center of this development, with innovative techniques being explored early for conditions such as spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD) [2–4]. In 2019, Zolgensma® (onasemnogene abeparvovec-xioi) –an adeno-associated virus vector-based gene therapy –received regulatory approval by the United States (US) Food and Drug Administration (FDA) for the treatment of 5q-linked SMA [5]. A year later, the European Medicines Agency (EMA) granted marketing authorization [6].
Although tremendously promising from a clinical point of view, the advent of gene therapies has also introduced challenges, in particular for payers. Extensive research, development, and manufacturing costs, in combination with small pools of eligible patients, means that the price of these novel health technologies often is very high [7, 8]. In addition, many gene therapies are administered only once and thus associated with high “one-off” prices. Zolgensma® (onasemnogene abeparvovec-xioi), for example, has a list price exceeding $2.1 million [9]. For these reasons, concerns have been raised regarding the suitability of current health policy systems, including health technology assessment (HTA) frameworks, in ensuring appropriate access to these therapeutic innovations while simultaneously safeguarding value for taxpayers’ money [10, 11].
With only a limited number of treatments currently approved globally, we are at the beginning of the gene therapy revolution [12]. However, hundreds of clinical trials of gene therapies targeting a wide range of conditions, including several monogenetic neuromuscular diseases, are currently ongoing [13]. Additionally, over time, as more and more diseases become amendable to gene therapy, the number of transformative, high-cost gene therapy products reaching the market is likely to increase considerably [14, 15]. This development has raised important questions concerning value, affordability, and access to gene therapies in a context of limited resources and constrained health budgets. The aim of this review is to provide a summary overview of current challenges and future perspectives of gene therapies for neuromuscular diseases from a health economic point of view.
Gene therapy
According to the US FDA, gene therapies are biological products that “mediate their effects by transcription or translation of transferred genetic material, or by specifically altering host (human) genetic sequences” [16]. The EMA defines a gene therapy as a biological medicinal product which has the following characteristics: “(a) it contains an active substance which contains or consists of a recombinant nucleic acid used in or administered to human beings with a view to regulating, repairing, replacing, adding or deleting a genetic sequence; (b) its therapeutic, prophylactic or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence. Gene therapy medicinal products shall not include vaccines against infectious diseases” [17].
Gene therapies consist of a vector or delivery formulation/system containing a genetic construct engineered to express a specific transgene. Two main categories of gene transfer exist: (1) in vivo, where the genetic modification of the cell takes place inside the body, and (2) ex vivo, where the cells are modified outside the body and then delivered back to the patient [18]. Additionally, there are four basic approaches employed in gene therapy: (1) gene addition (in which a new gene is added into desired cells to produce new proteins), (2) gene correction (which involves the use of gene-editing techniques to eliminate repeated or defective elements of a gene or to replace a defective or dysfunctional deoxyribonucleic acid [DNA] region), (3) gene silencing (which limits protein translation from the targeted messenger ribonucleic acid [mRNA]), and (4) cell elimination (through which cells are destroyed) [19]. A comparatively new technique to alter protein expression at the genome level is known as genome editing. Instead of introducing new genetic material into cells, genome editing introduces molecular tools to change the existing cell DNA [20], thereby enabling an entirely new modality for treatments based on precise modification of human genome sequences. At the time of writing, in vivo approaches based on adeno associated vectors (AAV) currently hold most promise for neuromuscular diseases [21].
The price of gene therapies
Gene therapies are typically very costly, out of grasp of most patients if not reimbursed (e.g., by the state or insurance provider). In fact, at a price point exceeding $2.1 million, Zolgensma® (onasemnogene abeparvovec-xioi) for SMA has been recognized as the “world’s most expensive drug” [22]. The high price per patient is due to a combination of both high fixed and high variable costs. A summary of key factors contributing to the high price point of gene therapies is provided below.
Factors contributing to the high price point of gene therapies
Accordingly, from a payer perspective, the challenge of absorbing new gene therapies is two-fold: (1) Ensuring cost-effectiveness, that is, value for money in relation to other approved health technologies, and (2) Ensuing affordability, that is, balancing budgets in the face of extraordinary costs. The second challenge is expected to become increasingly important as new gene therapies are developed and approved.
Concerning research and development, it is important to highlight that far from all costs associated with clinical programs of gene therapies are carried by pharmaceutical companies. A recent review of sponsorship and funding for gene therapy trials in the US [30] revealed that 10% of all trials were sponsored by the National Institutes of Health (NIH), 40% by the industry, 25% by hospitals, and 25% by universities. In total, 36% of all trials were funded solely by pharmaceutical companies, and 50% by academia or the NIH; however, the industry sponsored all phase III trials [30]. Accordingly, total research and development costs are commonly shared across multiple stakeholders, which should be considered in relation to decisions on commercial price points.
The current gene therapy landscape
On June 1, 2022, five gene therapies were approved for use in the US by the FDA [31], and 11 in the EU (including Norway, Iceland, and Liechtenstein) by the EMA [32]. Of these, Zolgensma® (onasemnogene abeparvovec-xioi) targets a neuromuscular disease, namely SMA. Yet, continued progress in the discovery of the genetic mechanisms underpinning many diseases have enabled the development of therapies targeting an increasing number of indications. Hundreds of clinical trials of gene therapies are planned or currently ongoing [13], and by 2025, the US FDA predicts that they will be approving 10 to 20 cell and gene therapy products per year [33].
The pipeline of gene therapies for neuromuscular diseases
A total of 28 interventional clinical trials of gene therapies for neuromuscular diseases were registered on ClinicalTrials.gov [34] up until and including 2021 (Fig. 1). Of these, 9 (32%) were phase I, 9 (32%) phase I/II, 1 (4%) phase II, 7 (25%) phase III, and 2 (7%) phase IV trials. In total, more than two thirds (68%) of all identified trials started in the last five years (i.e., 2017-2021), and 19 trials (including five starting between January 1 and May 31, 2022) were currently ongoing (i.e., “active, not recruiting”, “recruiting”, “enrolling by invitation”, and “not yet recruiting”) (Table 1). One trial starting in 2022 was suspended [35].
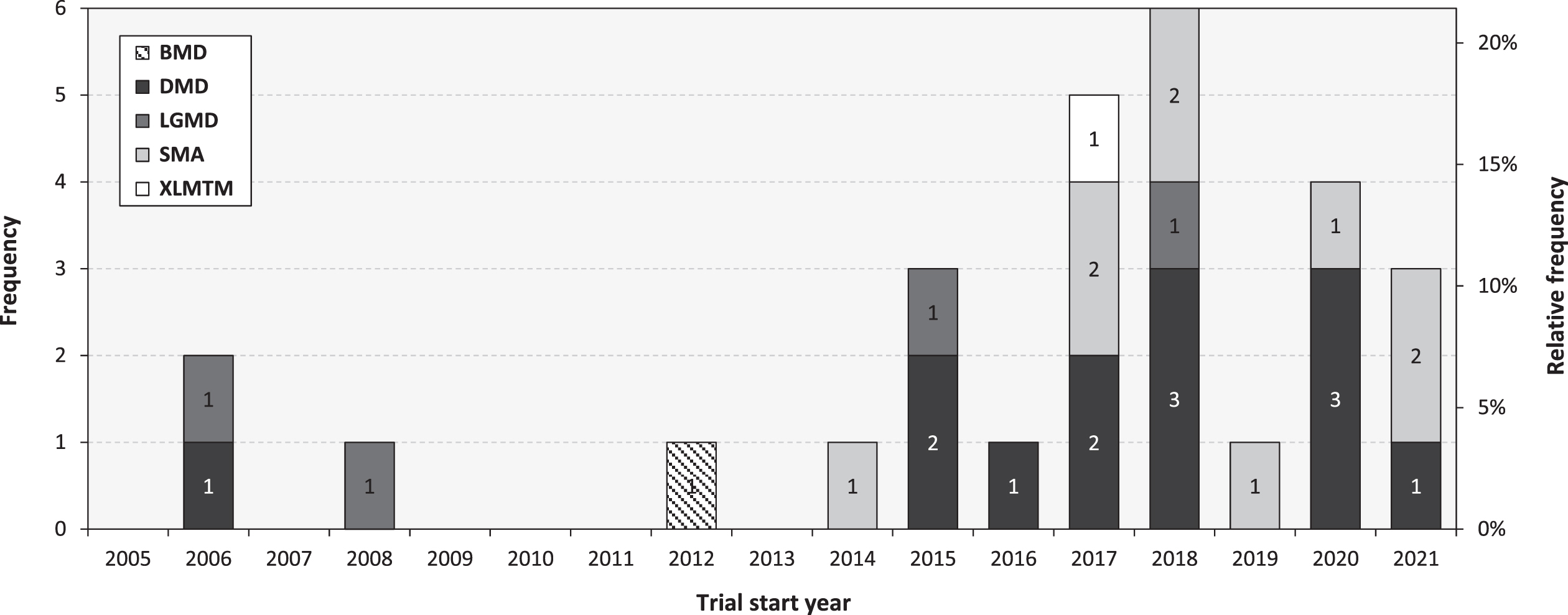
Interventional clinical trials of gene therapies for neuromuscular diseases. Note: The data shown was retrieved from ClinicalTrials.gov on June 1, 2022, based on a search of [(gene therapy) OR (gene treatment) OR (DNA therapy) OR (DNA treatment) OR (gene transfer)] AND (neuromuscular diseases), manually curated to exclude conditions other than neuromuscular diseases and interventions other than gene therapies. Only interventional trials with a specified phase were included for analysis. Becker Muscular Dystrophy (BMD). Duchenne Muscular Dystrophy (DMD). Limb Girdle Muscular Dystrophy (LGMD). Spinal Muscular Atrophy (SMA). X-Linked Myotubular Myopathy (XLMTM).
Ongoing interventional clinical trials of gene therapies of neuromuscular diseases
Note: The data shown was retrieved from ClinicalTrials.gov on June 14, 2022. Duchenne Muscular Dystrophy (DMD). Limb Girdle Muscular Dystrophy (LGMD). Spinal Muscular Atrophy (SMA). X-Linked Myotubular Myopathy (XLMTM). Argentina (AR). Australia (AU). Belgium (BE). Brazil (BR). Canada (CA). Denmark (DK). France (FR). Israel (IL). Italy (IT). Japan (JP). Russia (RU). Singapore (SG). South Korea (KR). Spain (ES). Switzerland (CH). Taiwan (TW). United Kingdom (UK). United States (US).
Regulatory approval of gene therapies
As human medicinal products, gene therapies are subject to rigorous evaluation to guarantee safety and efficacy with the primary objective to prevent harm [36]. In the US, human gene therapy products are regulated by the Center for Biologics Evaluation and Research (CBER) within the US FDA [37]. In the EU (including Iceland, Norway, and Liechtenstein), scientific regulatory assessment of advanced therapy medicines, including gene therapies, are performed by the Committee for Advanced Therapies (CAT) within the EMA [38].
Gene therapies commonly target rare diseases (defined as one in 2,000 in the EU [39] and about one in 1,650 in the US [40]). As such, in the regulatory approval process, they might be eligible for orphan drug designation programs. Incentives offered in the EU for medicines that have been granted an orphan designation by the European Commission includes scientific advice (referred to as “protocol assistance”), market exclusivity (i.e., ten years of protection from market competition with similar medicines with similar indications once they are approved), administrative and procedural assistance (for small and medium-sized enterprises), and fee reductions for regulatory activities [41]. In the US, the FDA has implemented similar incentives for orphan drugs, such as tax credits for qualified clinical trials, exemption from user fees, and market exclusivity schemes [42]. In general, these incentives have been put in place to help stimulate the development of treatments of rare diseases –so called “orphan drugs” due to their long-neglected position in the drug development landscape - based on principles of equity in access to healthcare, where individuals suffering from orphan diseases should be entitled to the same opportunity of receiving treatment as patients with more prevalent conditions [27, 28].
Additionally, many gene therapies are considered breakthrough innovations, offering major therapeutic advantage over existing treatment options. As such, they might be eligible for priority medicines (PRIME) designation, launched by the EMA in 2016 to enhance support for the development of medicines that target an unmet medical need [43]. PRIME is voluntary and based on enhanced interaction and early dialogue with developers of promising medicines to optimize development plans and speed up evaluation so these medicines can reach patients earlier. Two similar regulatory schemes, Breakthrough Therapy designation and the regenerative medicine advanced therapy (RMAT) designation, have been implemented by the US FDA, encompassing Fast Track designation features, intensive guidance on an efficient drug development program, and organizational commitment involving senior managers [44]. Further details and additional schemes provided by the FDA and EMA are reviewed in detailed elsewhere [45].
Considerations for regulatory approval of gene therapies
A recent systematic review concluded that the main driver for positive marketing authorization of human gene therapy products in the EU was clinical efficacy evidence, followed by safety considerations [46]. Commonly recognized inter-related challenges associated with obtaining adequate, high-quality clinical evidence of efficacy and safety of gene therapies are summarized in Table 2. Taken together, these issues means that evidence of efficacy and safety of gene therapies, in particular long-term data, often is uncertain at time of regulatory approval and HTA.
Common challenges characterizing gene therapy trials
Note: Duchenne Muscular Dystrophy (DMD).
Health technology assessment of gene therapies
Health technology assessment (HTA) has been defined as “the systematic evaluation of the properties, effects, and/or other impacts of healthcare technology” [54]. In this context, a healthcare technology is “any intervention that may be used to promote health, to prevent, diagnose or treat disease or for rehabilitation or long-term care” [55], encompassing also advanced therapy medicinal products, such as gene therapies. The main purpose of HTA is to inform policymaking, including pricing and reimbursement decisions, for existing and new healthcare technologies [56].
Although centralized procedures for marketing authorization exists in some regions (e.g., the EU), HTA of new medicines, including reimbursement of gene therapies, are made at the national level [57]. These decisions usually consider evidence of cost-effectiveness from economic evaluations. In such analyses, generally performed using mathematical disease models, the incremental cost of the new treatment (A) versus the comparator treatment (B), C A -C B =ΔC, is put in relation to the incremental benefit, E A -E B =ΔE. Costs include those related to the treatment and disease carried by the payer, although in some settings additional elements may also be included, including informal care and absenteeism and presenteeism from work from a social perspective [58]. Benefits are usually expressed in terms of quality-adjusted life-years (QALYs), a measure incorporating both the quality and quantity of life, created by multiplying every life year with a utility weight (ranging between 0 and 1) reflecting health-related quality of life [59]. The time horizon for the assessment varies; however, for gene therapies, and other interventions with expected long-term benefits, the full lifetime of the patient is usually considered relevant [52, 60] (across which benefits and costs are measured and discounted to present values [61]).
In the case above, the outcome of the economic evaluation is expressed as the incremental cost-effectiveness ratio (ICER), ΔC/ΔE, or “cost per QALY gained”. To determine if the treatment is cost-effective versus the comparator, the estimated ICER is subsequently tested against a monetary societal willingness-to-pay (WTP) threshold for a QALY [59]. In this way, it is possible to compare the cost-effectiveness of different health technologies, even across therapeutical areas, to decide which represents the best value for money.
Similar to assessment informing regulatory approval, the lack of robust efficacy and safety data for many gene therapies is a challenge also for HTA. Moreover, because of their very high prices, gene therapies are generally not found to be cost-effective when tested against conventional WTP thresholds [60, 62]. For these reasons, voices have been raised to also considered other factors in the reimbursement appraisal of gene therapies to facilitate patient access, including but not limited to the potential additional value of (1) curing a disease (as opposed to chronic treatment administration), (2) treating a very severe disease (as opposed to a nonfatal, less debilitating illness), (3) treating a rare disease (as opposed to a common condition), and (4) treating a disease with a very high unmet medical need (as opposed to an illness with many efficacious treatment options). Interestingly, there appears to be conflicting evidence of preferences for a higher WTP for a QALY based on these considerations among the general public [63–67]. That being said, more research is needed to further delineate preferences for these topics.
A separate, but closely related issue of value assessments of gene therapies concerns affordability, commonly referring to payers’ potential to absorb new health technologies at (ideally value-based) price points within current budget constraints [8]. Indeed, as previously described, most gene therapies are expected to be marketed at high one-off prices. Therefore, the reimbursement of a new gene therapy is likely to put significant pressure on the payer’s budget [68]. For example, DeMartino et al. [69] concluded that the reimbursement of a gene therapy for sickle cell disease would likely present affordability challenges to several Medicaid health plans, and Alhakamy et al. [17] estimated that the total cost of gene therapies for only 10% of US patients with genetic diseases (accounting to about 1% of the total population, according to the authors) could be as large as $3 trillion, roughly 75% of the total national healthcare expenditure in 2020 [70].
Payment models for gene therapies
To help realize the potential of these innovative medicines, and manage risks across involved stakeholders, a myriad of payment models of various complexity has been explored to alleviate the two primary challenges of gene therapies from a HTA perspective, namely extraordinary high one-time costs and uncertain clinical evidence. These payment models are frequently implemented in parallel with conditional approval, or coverage with evidence development (CED) schemes, in which the pharmaceutical company is mandated to continuously collect data on effectiveness and safety to inform future reimbursement decisions [68]. A summary of two of the most common payment models is presented below (for a more in-depth account, readers are referred elsewhere [7, 8]).
Annuity and payment-by-performance payment models
A recent systematic review [68] of the use of novel payment mechanisms for gene therapies in France, Germany, Italy, Spain, the UK, and the US found that many countries commonly employ CED schemes, as well as payment-by-performance models, to fund these medicines. For example, in Germany, reimbursement of Zolgensma® (onasemnogene abeparvovec) was approved as part of a payment-by-performance “claw-back” model, encompassing refunds of up to 100% based on patient-relevant outcomes. Moreover, in the US, reimbursement of Zolgensma® (onasemnogene abeparvovec) includes different annuity models with and without payment-by-performance rebates [68].
Patient access to gene therapies
Historically, reimbursement of orphan drugs, including treatments of rare neuromuscular diseases, have varied markedly both within and across countries. Restrictions in reimbursement of orphan drugs have been reported in, for example, Central and Eastern European countries [71], Italy [72], Spain [73], and Turkey [74], as well as other settings [75]. Recently, Ward et al. [76] compared reimbursement status of marketed orphan drugs in 18 European countries, as well as Canada, between January 1, 2015, and March 31, 2020. The authors found that the proportion of orphan drugs with reimbursement ranged from 84% in Germany to 23% in Croatia, with the majority of countries reimbursing < 50% of available treatments of rare diseases. In addition, striking variability was noted in access across provinces in Canada, ranging from 32% in Ontario to 3% on Prince Edward Island [76].
Given their extraordinarily high prices and uncertain efficacy and safety profiles, ensuring equality in access is expected to be a challenge also for gene therapies. In a recent systematic review, Margaretos et al. [77] studied the coverage of Zolgensma® (onasemnogene abeparvovec-xioi) across 17 large US private insurers (covering approximately 150 million people, equal to about 60% of the private health insurance market) as of April 2020. The authors found that 16 plans reimbursed Zolgensma® (onasemnogene abeparvovec-xioi), but that they all imposed restrictions in coverage. For example, four plans reserved coverage for patients with SMA type I and two plans for SMA type II, while the remaining ten did not specify SMA type in their policies (per the FDA label). Other factors that varied included number of copies of the SMN2 gene, patient age, and prescriber requirement [77].
summary and future perspectives of gene therapy for neuromuscular diseases
The first parts of this review aimed to provide an overview of key characteristics common to many gene therapies. In summary, these are: Highly efficacious, potentially curative; Targets rare diseases with high unmet medical need; Complex manufacturing and delivery processes; Single (one-off) administration; Uncertain efficacy and safety, in particular in the long-term; Very high prices; and Few currently marketed, but extensive development pipeline, also encompassing several monogenetic neuromuscular diseases.
As illustrated in this review, these features are associated with three key challenges for decision-makers when meeting public demands for innovative, high-cost therapies: (1) evaluating risk-benefit ratios, (2) evaluating cost-effectiveness, (3) ensuring affordability and health system sustainability.
Regarding evaluation of risk-benefit ratios, it is clear that few gene therapies for neuromuscular diseases will have adequate evidence of absolute and relative efficacy and safety at the time of seeking regulatory approval. Agencies such as the EMA would thus be expected to continue provide conditional approval mandating post-authorization efficacy studies (PAESs) and post-authorization safety studies (PASSs) to ensure that the benefit and harm profiles recorded within the clinical program also are replicated to a satisfactory degree in a real-world setting. Initiatives that could help manage this development include the establishment of disease registries [78–80] facilitating patient identification and recruitment to rare disease research, as well as multi-national data collection infrastructure recording real-world patient outcomes at pre-defined milestones in relation to the date of treatment initiation. A welcome addition in this context is the recently published Registry Evaluation and Quality Standards Tool (REQueST) [81], a set of criteria developed in partnership with several European HTA bodies to help assess the quality of existing registries that could be utilized for real-world evidence generation.
Regarding the evaluation of cost-effectiveness, it is clear that payers will struggle with assessing value for money of many gene therapies for neuromuscular diseases due to lack of long-term efficacy and safety data. It is also clear that few gene therapies will meet conventional WTP thresholds. Arguments have been made for higher WTP thresholds for gene therapies [8, 82–85]. However, as noted, there appears to be inconclusive support among the general public for such special considerations [62–66]. As a result, reimbursing these treatments will ultimately lead to inefficiencies, as more health could have been produced at the same or lower cost with other health technologies [58].
Regarding affordability and sustainability, it is clear that reimbursement of gene therapies is likely to have a non-trivial impact on payers’ budgets, in particular as the current pipeline is brought to market over the coming decades. Although individually rare, recent estimates indicate that there are more than 10,000 distinct rare diseases [86], affecting an estimated 30 million people in the EU and US, respectively [87, 88]. Of these, more than 780 constitute monogenetic neuromuscular diseases [89]. As a result, the total number of patients treated with gene therapies across therapeutic areas in the future could be quite sizable. At the population level, the budget impact from funding a gene therapy would be expected to peak in the time period following reimbursement approval, as additional cases would be relatively rare given the low disease incidence. Any scheme implemented to manage the budget impact of gene therapies should ideally also be able to manage such foreseeable temporal shocks.
Taken together, these three challenges associated with gene therapies are likely to restrict access for neuromuscular patients in many parts of the World, in particular less-developed countries. This should not come as a surprise. In fact, although it is true that “...one-off curative gene therapies for diseases such as sickle cell disease hold particular promise for patients in low- and middle-income countries, where the long-term management of the disease is particularly challenging due to resource-strained healthcare systems" [90], it is equally true that the money spent on, for example, Zolgensma® (onasemnogene abeparvovec-xioi) saving one patient in Africa instead could have been used to save or improved the lives of many. To be specific, saving one life in Africa using insecticide-treated bed nets (at $5 apiece) to prevent deaths due to malaria has been estimated to cost about $4,500 (details provided here [91]). The harsh reality is that the money spent on saving one patient with SMA in Africa using Zolgensma® (onasemnogene abeparvovec-xioi) thus could have been used to save more than 450 lives by purchasing and delivering cheap nets. This is obviously an oversimplification, but it illustrates the point. Additionally, gene therapies are expensive also because of their resource-intensive manufacturing processes [24–26]. As a result, the gene therapy breakthrough would not be expected to progress through the poorer regions of the World for many years, even decades, because of lack of appropriate manufacturing, processing, and delivery infrastructure, as well as human resources, experience, and expertise. There are, however, other reasons not related to costs for why gene therapies might not be within reach of all patients globally. Gene therapies frequently target rare or ultra-rare disorders, and some smaller countries might not have a sufficient number of patients to justify and support a multidisciplinary national specialist treatment center with sufficient patient exposure and experience to administered high cost, cutting-edge health technologies. Accordingly, geographical coverage of each and every eligible patient is probably neither efficient, nor feasible.
In terms of managing affordability, payment-by-performance annuity payment models appears to have several attractive features fitting for gene therapy products. For example, a problem with single-administration therapies compared with conventional medicines is that it is not possible to terminate treatment in case of low (or fully absent) effectiveness. An annuity payment model linked to observed treatment outcomes might be a reasonable path forward, since it facilitates budget management (by spreading out costs over time) while simultaneously allowing payers to terminate payment in case of poor treatment performance. Mandating follow-up of real-world outcomes as part of such a scheme will also help generate much-needed data on long-term therapy effectiveness and safety. Finally, payment-by-performance payment models create natural incentives for manufacturers to not overestimate long-term treatment benefits in economic evaluations.
Real-world evidence will likely play a prominent role in the future of gene therapies [8, 15], from contextualizing single-arm trial efficacy data via external comparators, to monitoring of post-launch effectiveness and safety. This has been recognized by, for example, the National Institute for Health and Care Excellence (NICE) in the UK, recently launching a real-world evidence framework aiming to clearly describe best-practices for the planning, conduct, and reporting of real-world evidence studies, improve the transparency and quality of real-world evidence, improve trust in real-world evidence studies, and ensure real-world evidence is used where it helps to reduce uncertainties, improve recommendations, and speed up access of patients to new effective interventions [92]. A similarly themed framework has been launched in the US by the FDA [93]. In the case of neuromuscular diseases, of which many are rare, this is a positive development considering general challenges in identifying and recruiting patients to research in controlled settings [47, 48]. Yet, additional data collection platforms and similar infrastructure are undoubtedly needed to ensure reliable recording of relevant real-world gene therapy outcomes at a global scale. Efforts to establish such networks must probably intensify in coming years to avoid delaying future gene therapy development and evaluation. More research is also needed to help develop fit for purpose outcome measures to ensure that treatment benefits, if any, can be meaningfully measured and interpreted [51].
In conclusion, gene therapy for neuromuscular diseases holds immense clinical potential, but also present complex challenges. The key question is how to balance incentives for research and development of orphan drugs while simultaneously meeting demands for access to approved therapies, safeguarding value for taxpayers’ money, and ensuring affordability and sustainability of the health system. Yet, in most settings at current prices, gene therapy would not be considered representing good value for money. Alas, similar to other extraordinarily expensive products, there is thus a significant risk that gene therapy indeed becomes “a prime example of healthcare inequity” [94]. Payment-by-performance annuity payment models could help mitigate challenges with affordability of the gene therapy revolution, the clinical, social, and economic impact of which is likely to be quantified and described predominantly using real-world evidence.
CONFLICTS OF INTEREST
None.