
Abstract
Background:
Glycogenosis type 11 or deficiency in lactate dehydrogenase A (LDHA) (OMIM: 612933) is an ultra-rare condition of perturbed glycogen metabolism, first described in 1980 in a Japanese patient, and quite rare outside Japan. There are very few cases described in the literature and there is limited awareness of this condition that can easily be misdiagnosed or remain undiagnosed.
Objective:
To report on an ultra-rare form of glycogenosis stressing the association with cutaneous features and raise awareness for this rare condition. We report a novel pathogenic variant and aim to contribute to the understanding of the myopathophysiological mechanisms of disease by proteomics.
Methods:
We report the clinical, histopathological, magnetic resonance, genetic and proteomic features of a 19-year-old male of North African background that presented from infancy episodes of muscle pain, contractures and high CK levels immediately after moderate to high-intensity exercise. The patient was followed for several years prior to our observation due to severe acne that involved mostly the back and was resistant to treatment.
Results:
The phenotype of this patient is similar to the ones previously described with muscle and skin involvement. The MRI functional studies, interrupted at 2’30’’ due to muscle pain, allowed us to confirm the presence of a metabolic disturbance of the muscles. The histological features of the muscle were quite subtle consisting mainly in rare subsarcolemmal vacuoles also identified at ultra-structural level. Immunostaining with the antibody targeting LDHA showed intracytoplasmic aggregates of the protein unlike the normal control that presented a diffuse staining. Exome analysis revealed a novel bi-allelic frameshifting deletion in exon 7 of the LDHA gene; c.766_767delGT. Proteomic findings accord with loss of functional LDHA and provide significant insights into the pathobiochemistry of the disease.
Conclusions:
Our combined data expand the current genetic landscape of LDHA-related disease, confirms the concept of a metabolic-driven vacuolar myopathy and provides insights into the biochemical nature of myopathology.
Keywords
Introduction
Glycogenosis type 11 or deficiency in lactate dehydrogenase A (LDHA) (OMIM: 612933) is an ultra-rare condition of impaired glycogen metabolism, inherited in autosomal recessive fashion, and first described in 1980 in a Japanese patient.1,2 The disease is caused by mutations in the LDHA gene.
The clinical features include exercise intolerance due to muscle pain and stiffness during intense exercise. Later, signs and symptoms such as cutaneous lesions and uterine stiffness were added to the phenotype.3–6 LDHA is localized on chromosome 11 and encodes lactate dehydrogenase (LDH) subunit A which is expressed in skeletal muscle and involved in the conversion of lactate and pyruvate. Other isoforms of LDH are LDHB and LDHC, the former expressed in cardiac muscle and the latter in the testis.
We report the clinical, histopathological, magnetic resonance and genetic features of a 19-year-old male of North African background that harbor's a novel bi-allelic mutation in the gene LDHA. Characterization of myopathology also included determination of the proteomic signature of LDHA-mutant muscle derived from this patient.
Materials and methods
Muscle biopsy
The patient gave informed consent for muscle biopsy, genetic analysis and reuse of biological samples for research. The muscle biopsy was an open muscle biopsy obtained from the deltoid muscle. It was snap frozen in isopentane cooled in liquid nitrogen for histoenzymology and immunohistochemistry and included in epon for electron microscopy.7,8
We performed immunostaining with antibodies against the membrane-associated proteins: Dystrophin (Novocastra: NCL-DYS1, NCL-DYS2, NCL-DYS3), Sarcoglycans (Novocastra: NCL-g-SARC, NCL-b-SARC, NCL-a-SARC, NCL-d-SARC), Dysferlin (Novocastra: NCL-Hamlet), Laminin α2 (Novocastra: NCL-MEROSINE), Caveolin-3 (GeneTex: GTX109650), β-Spectrin (Novocastra: NCL-SPEC1)). Antibodies against Telethonin (Santa Cruz: sc-25327), LDHA (Santa-Cruz: sc133123) were used in addition to antibodies used to study autophagy: anti-LC3 (Sigma: L7543) & anti-LAMP2 (abcam: ab25223)) and such against C5b-9 (Dako: M0777 clone aE11) and MHC class I (Dako: M0736 clone W6/32).
Muscle MRI
Quantitative MRI was carried out at 3 T (PRISMATM, Siemens Healthineers) using the body coil for radiofrequency transmission and a combination of two 18-channel flexible coils placed on the thighs and the chest and one 32-channel spine coil for signal reception. A multi-echo 3D gradient echo (GRE) sequence, a multi-spin echo (MSE) and the MRF T1-FF sequences were performed on the thighs and legs. The reference 3-point Dixon method was applied to the multi-echo 3D-GRE acquisition to assess the intramuscular fat fraction (FF). T2-weighted short-tau inversion recovery was acquired on the thigh and leg to assess muscle oedema. Muscle lesions were categorized according to the Mercuri grading score.
An in-house dynamic assessment protocol based on a plantar flexion against progressively increased resistance and intermittent 31P spectroscopy aimed at evaluating the exercise intolerance of the patient was performed. It is noteworthy to mention the despite good patient participation to the exercise this one had to be interrupted prematurely due to increasing muscle pain.
Genetics
DNA extracted from circulating white blood cells was prepared for whole exome sequencing. The NEBNext Ultra II DNA Library preparation kit for Illumina 2 was used, and the capture kit was the Twist Human core Enrichment System (Twist Bioscience) associated with the IntraGen Custom. Exome sequencing used the NovaSeq Sequencer with a maximum read length of 2 × 100 bp. Sanger sequencing confirmed the results. The variants description uses the HGVS nomenclature. 9
Proteomic profiling
To elucidate biochemical changes taking place upon loss of functional LDHA in skeletal muscle, we performed unbiased proteomic profiling making use of a data-independent-acquisition approach as published previously. 10
Results
Case report
The patient is a 19 years old male student, born to consanguineous healthy parents (first degree cousins) of North African origin. Presents from infancy episodes of muscle pain, muscle contractures and high CK levels that occurred immediately after exercise of moderate to high intensity and with repetitive movements. These episodes would last up to one week, and gentle movements of the affected segments relieved the pain. There was reference to one episode of dark urine without renal involvement. Outside these episodes, the patient was active and athletic. There were no complaints of muscle weakness or atrophy, no second wind phenomenon, no myotonia and no exposure to drugs or medication. The dermatologists followed the patient for several years prior to our observation due to severe acne that involved mostly the back and was resistant to different treatments. The neurological and general examinations were normal except for the presence of nodular and purulent cystic lesions and confluent infiltrates in the trunk associated with pronounced scarring (Figure 1). There was no family history of muscle diseases. The proband has two healthy sisters. The electromyogram showed a diffuse pseudo-neurogenic pattern compatible with chronic myogenic damage. Lactate and pyruvate blood levels at rest were increased at respectively 2.4 mM and 204 mM. The serum level of CK at rest was 2580 IU/L and rose to 6000 IU/L after a football game. The cardiac and respiratory evaluation was normal.
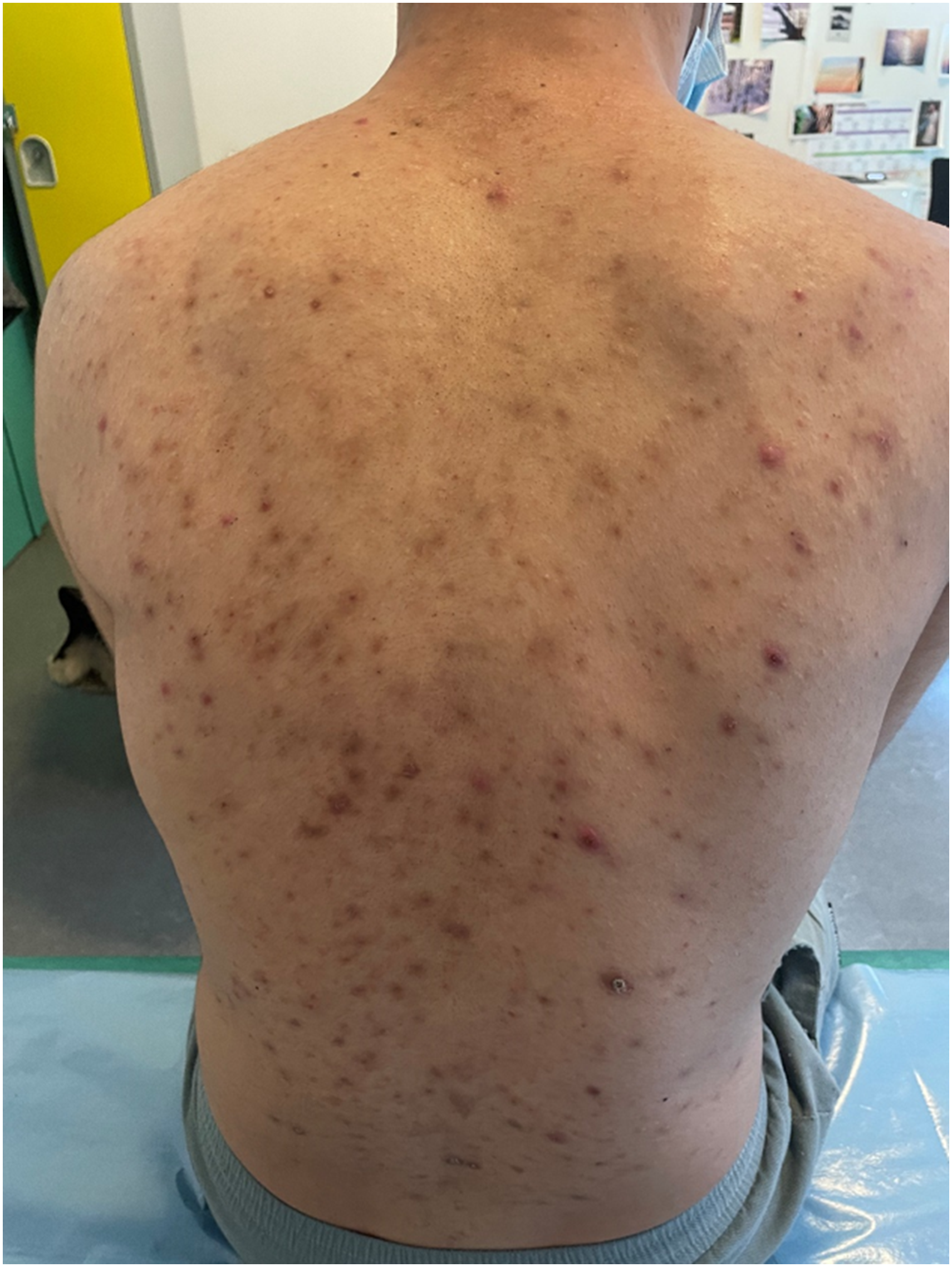
Image of the dorsal region of the patient showing the skin lesions in particular the pronounced scarring.
Whole body muscle MRI
Whole body muscle MRI was unremarkable on T1 (Figure 2) and T2. The functional studies interrupted at 2’30’’ due to muscle pain did not allow the quantification of the energetic depletion of the muscle. Spectroscopy did not reveal abnormal phosphocreatine depletion or unusual acidification of the muscle tissue. Even if dynamic assessment did not reveal any specific abnormality, it could contribute to the objectification of exercise intolerance.
Shows representative slices (T1-weighted images) ranging from the scapular belt to the legs. No significant alterations were observed at the time of the exam.
Muscle biopsy findings
On hematoxylin-eosin (H&E), myopathological findings were quite subtle however, we could observe abnormal variability of the fiber size with the presence of round or polygonal atrophic fibers scattered through the biopsy. It was possible to identify occasional fibers with subsarcolemmal vacuoles optically empty. PAS stain showed some intensely stained fibres, and some of the subsarcolemmal vacuoles had a PAS-positive content. (Figure 3) All PAS-positive material was digested on PAS-Diastase. The oxidative stains, particularly the DPNH, showed a moth-eaten aspect of muscle fibres. Acid phosphatase staining did not reveal any abnormal staining. Immunostaining with the antibody against LDHA showed intracytoplasmic aggregates of the protein unlike the normal control that presented a diffuse staining (Figure 3).
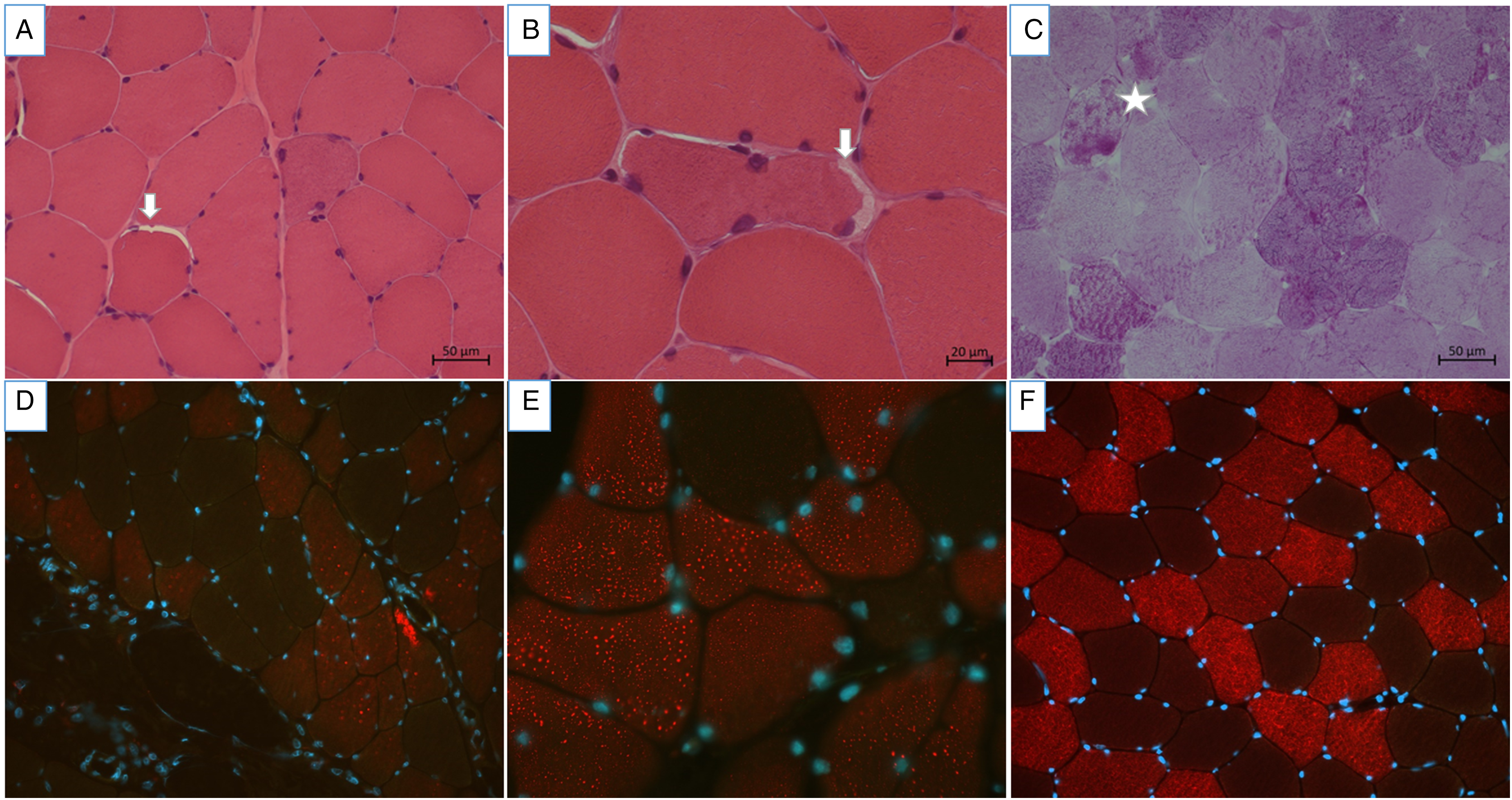
A and B H&E at 20x and 40 x respectively showing rare sub-sarcolemmal empty vacuoles (arrows). C. PAS at x 20 showing one of the rare fibers were there was a discrete increase in the content of PAS positive material (star). Immunostaining with the antibody against LDHA showing (D (x25); E (x63)) intracytoplasmic aggregates of the protein in the patient. (F (x25)) Control showing a diffuse staining.
Immunolabelling with the antibodies directed against the sarcolemma-associated proteins was normal (data not shown) with no evidence of ectopic protein deposition (no particular sarcolemmal features). The immunostaining with anti-LC3 and anti-LAMP2 antibodies was normal (data not shown). Immunostaining with antibodies against HLA 1 and C5b-9 showed some small groups of fibers with sarcolemmal and sarcoplasmic punctate HLA1 stain and only 3 fibers showed sarcolemmal punctate stain with C5b-9. The antibody against LDHA was used to demonstrate that the protein aggregated in the cytoplasm of muscle cells. (Figure 3).
On electron microscopy, there was an increased content of free glycogen, localized either in the sub-sarcolemmal region or in between the myofibrils. There were rare sub-sarcolemmal vacuoles surrounded by membrane had a content reminiscent of glycogen granules. In addition, small autophagic vacuoles located in the subsarcolemmal region and within the cytoplasm were identified (Figure 4).
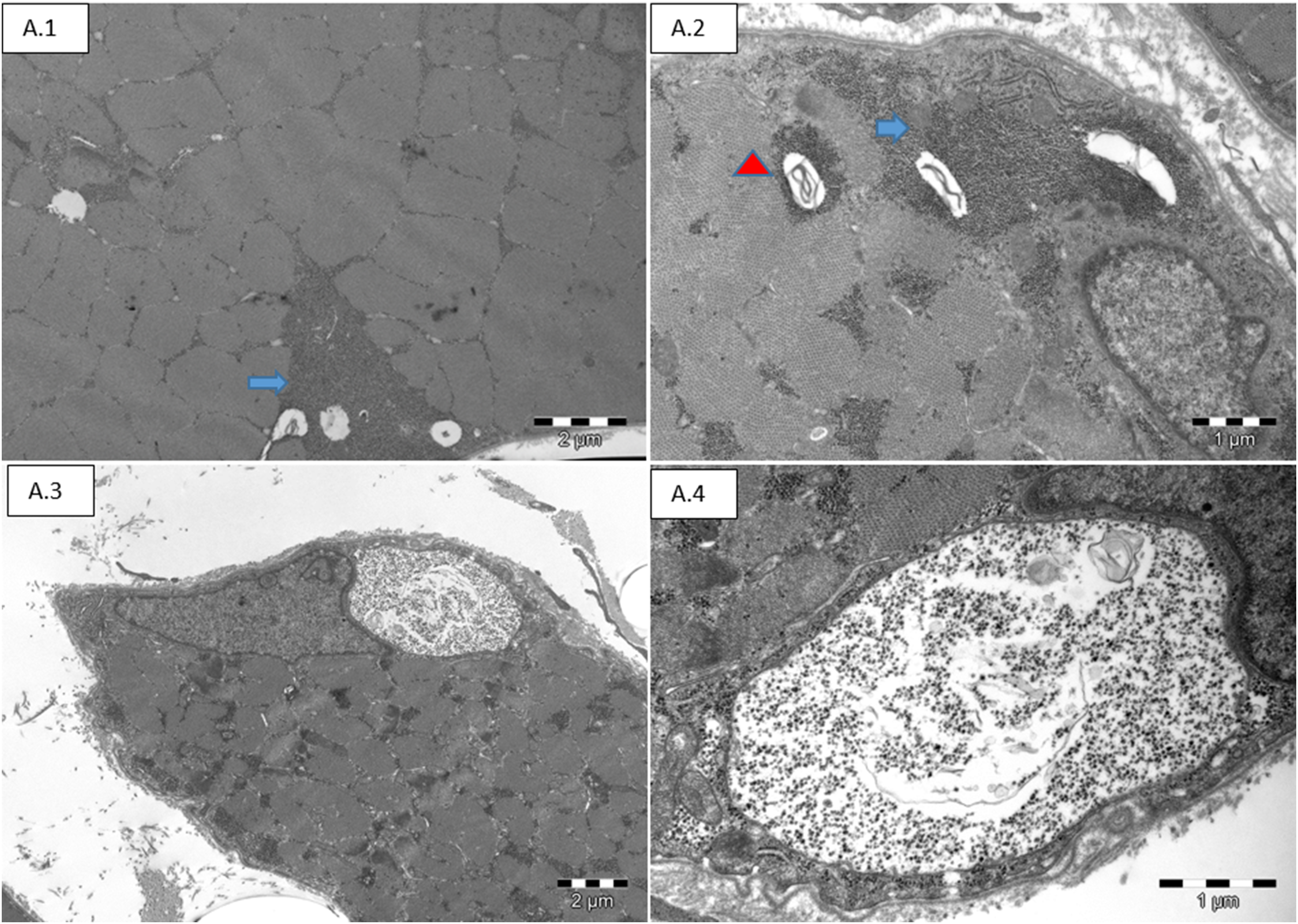
Electron microscopy: A.1 and A.2 show accumulated non-membrane-bounded glycogen granules in the sub-sarcolemmal area (arrow). A.2 Autophagic vacuoles with myelin-like bodies located mainly in the sub-sarcolemmal areas (arrowhead). A.4 and A.5 shows subsarcolemmal accumulations of glycogen granules surrounded by single-membrane.
Genetics
Exome analysis revealed a bi-allelic frameshifting deletion in exon 7 of the LDHA gene; c.766_767delGT, responsible for the appearance of a premature stop codon upstream of the last exon p. (Val256Serfs*12). This variation, classified 5 according to HGVS, was not previously reported in the literature. The appearance of the premature stop codon suggests a total deficiency of the LDHA protein.
Segregation studies have shown that both parents were carriers of the deletion in exon 7 of the LDHA gene.
Proteomic signature of LDHA-mutant muscle biopsy
Proteomic profiling on whole protein extracts from deltoid muscle (Figure 5A) enabled the robust quantification of 2451 proteins (Figure 5B). Of those, 254 proteins were significantly increased while 50 proteins were significantly decreased (Figure 5C). Each of these dysregulated proteins (12.4% of the total quantified proteins) was quantified based on at least two unique peptides. In line with our immunostaining data, mass spectrometry-based protein quantification showed abundance in patient-derived muscle, however with reduced level compared to control muscles (Figure 5D). This quantification is based on two unique tryptic peptides for LDHA in the patient (Figure 5E). Biological processes affected by increased proteins include cellular oxidant detoxification along with hydrogen peroxide catabolic processes organization of actin cytoskeleton and innate immune response accompanied by complement activation (classical pathway) (Figure 5F). Along this line, biological functions affected by the proteins with increased abundance include antioxidant, peroxidase, and oxygen transporter activity in addition to actin filament binding and extracellular matrix structural constituent (Figure 5F). Subcellular structures and organelles affected by the increased proteins include extracellular exosome and cytosol, Endoplasmic/ Sarcoplasmic Reticulum lumen, vesicles, and cytoskeleton (Figure 5F). Of note, biological processes modulated by the decreased proteins include exosomal secretion, epidermis development, intermediate filament organization, myofibril assembly along with muscle filament sliding (Figure 5F). Molecular functions affected by the decreased proteins include glyceraldehyde-3-phosphate dehydrogenase (NAD+) (non-phosphorylating) and aldehyde dehydrogenase (NAD) activity along with oxidoreductase activity (acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor) as well as NADH dehydrogenase activity and myosin phosphatase activity (Figure 5F). Subcellular structures and organelles affected by proteins with decreased abundance include extracellular exosome, cytosol/ sarcoplasm, mitochondrion, Z disc and peroxisomes (Figure 5F).
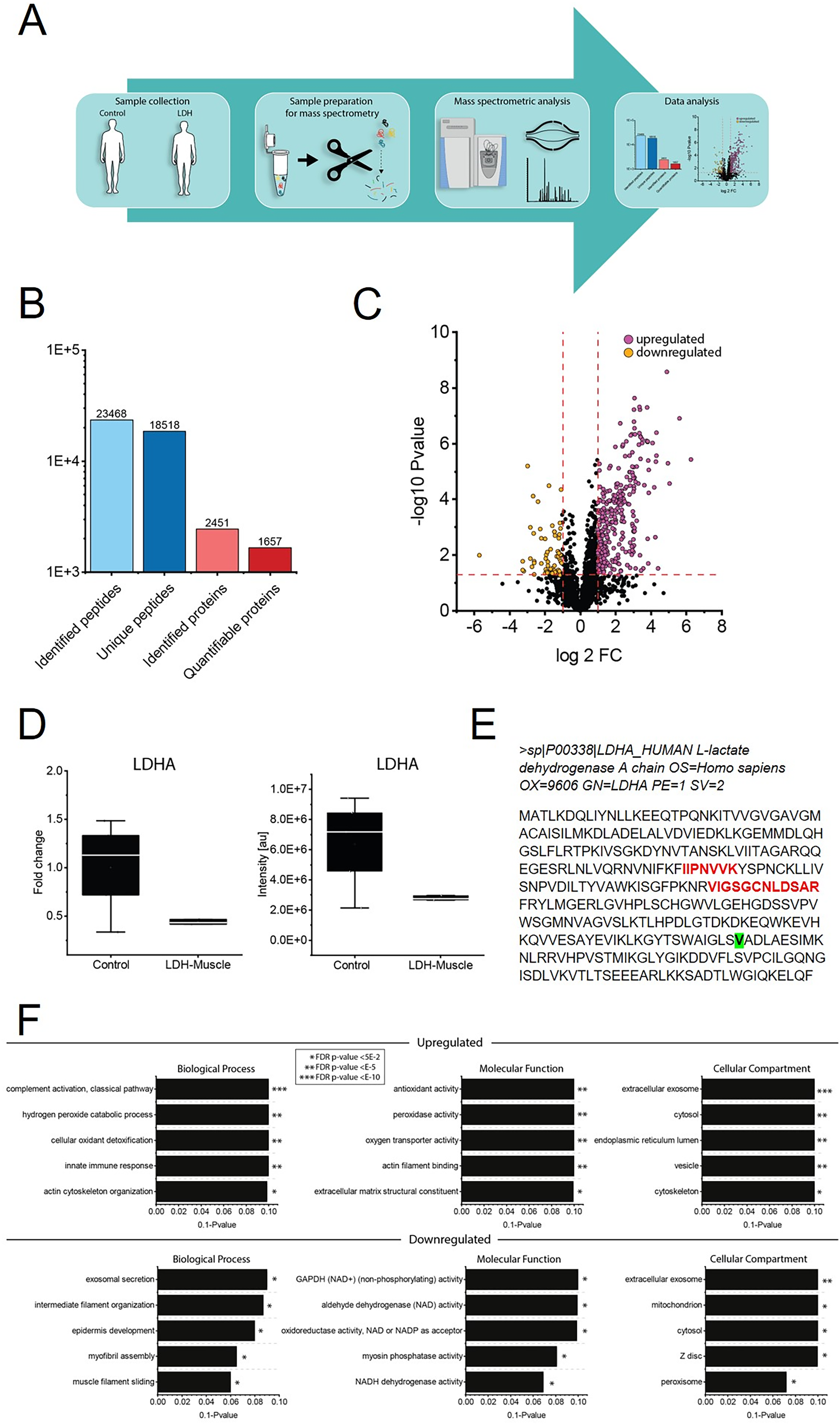
Proteomic studies on whole protein extract of LDHA-mutant muscle. (A) Schematic representation of the applied workflow. (B) Overall statistics of LDHA skeletal muscle proteomics. (C) Volcano plot showing proteins identified as being decreased (orange dots) and increased (purple dots) in PTHS patient-derived muscle. (D) Box-plot showing the profound (non-significant) reduction of LDHA in muscle protein extract derived from the patient compared to abundances in control muscle (left panel). Same illustration of reduced LDHA level in the patient based on intensities [au] is shown in the right panel. (E) Illustration of the two unique tryptic peptides (labelled in red) on which LDHA quantification is based in muscle protein extract of the patient. The amino acid (Valin) affected by the homozygous variant is highlighted in green in the amino acid sequence. (F) Results of GO-term based data analysis indicating activation of complement cascade, oxidative stress burden along with altered nicotinamide adenine dinucleotide metabolism as well as perturbed cytoskeleton impacting on muscle filament sliding.
Discussion
We report on a novel pathogenic variant occurring affecting the LDHA gene that codes for the protein lactate dehydrogenase A (LDHA). Lactate dehydrogenase A (LDHA) is an enzyme that catalyses the conversion of L-lactate and NAD to pyruvate and NADH in the final step of anaerobic glycolysis.
LDHA is expressed mostly in skeletal muscle and its deficiency is associated with exertional myoglobinuria. 1 LDHA gene mutations result in the production of an abnormal lactate dehydrogenase-A subunit unable to bind to the other subunits to form the lactate dehydrogenase enzyme. A lack of functional subunits reduces the amount of enzyme that is formed, mostly affecting skeletal muscles. As a result, glycogen is not broken down efficiently, leading to decreased energy.
Lactate dehydrogenase deficiency is a rare disorder. However, the exact frequency of pathogenic LDHA variants in the population (leading to manifestation of a neurological disease) is unknown. In a study from 1984, in Japan, this condition was estimated to affect around 1 in 1 million individuals. The authors screened 3776 blood samples and calculated the frequencies of homozygotes with LDHA subunit deficiency those were estimated as 14.20 x I0−7. 11
The clinical phenotype of our patient with rhabdomyolysis and muscle pain related with effort together with the severe acneiform cutaneous lesions and the biochemical aspects with high lactate and pyruvate levels at rest are compatible with what was previously described in the literature in association with pathogenic variants on the LDHA gene.
Muscle MRI was not contributive for the diagnostic workup but, even if dynamic assessment did not reveal any specific abnormality, it contributed to the objectification of exercise intolerance.
The only literature report describing the features of the muscle biopsy in these patients is quite limited in its description referring only that PAS staining indicated normal glycogen content. 12 In the muscle biopsy analysis of our patient, we have put in evidence several morphological features, marking the first-time complete description of histological/ myopathological characteristics in this context. The H&E staining revealed subtle yet significant findings notably, occasional fibers with sub-sarcolemmal vacuoles. Unlike the previously described, the PAS staining highlighted intensely stained fibers, with some sub-sarcolemmal vacuoles containing PAS-positive content. Immunostaining with antibodies revealing abnormalities in autophagy pathways, along with acid phosphatase staining, did not exhibit any aberrant patterns, differentiating this case from other glycogenosis myopathies like Pompe or Danon disease. Latter one is also characterized by perturbed mitochondrial function. 12 Immunostaining with LDHA antibodies demonstrated intracytoplasmic aggregates of the protein, contrasting with the diffuse staining observed in normal control. This microscopic finding not only accords with the finding of small vacuoles but also supports the pathogenic character of the identified variant: the bi-allelic frameshifting deletion fond in our patient leads to the creation of a premature stop codon and – based on our immunostaining data – most probably to the production of a truncated protein making it prone to aggregation. Thus, our combined immunostaining and proteomic data indicate that the variant is not a null but will result in a short protein and one might speculate that reduced level may result from cellular attempts of degradation of the aggregated protein.
Immunostaining with antibodies against HLA 1 and C5b-9 although unspecific correlate with the activation of the complement system and initiation of immune response as part of the underlying myopathology. Electron microscopy, proved useful in determining the nature of the myopathy by revealing an increased content of free glycogen, and confirming that LDHA deficiency is a vacuolar myopathy. These findings collectively contribute novel insights into the histological and ultrastructural features associated with the condition, providing a foundation for further understanding the pathological mechanisms at play in this unique case.
Moreover, the results of our proteomic profiling approach on the muscle biopsy strongly support the concept of a pathophysiological impact of loss of functional LDHA (and thus also confirm pathogenicity of the new variant) in skeletal muscle: decreased proteins impact on molecular functions regularly controlled by LDHA such as glyceraldehyde-3-phosphate dehydrogenase (NAD+) (non-phosphorylating) and aldehyde dehydrogenase (NAD) activities along with oxidoreductase activity (acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor) as well as NADH dehydrogenase activity. An impact of these perturbed activities on cellular redox status is clearly indicated by the function of a variety of proteins presenting with increased abundance modulating peroxidase function and antioxidant properties. This may reflect a cellular strategy antagonizing the oxidative stress burden arising from loss of functional LDHA. Representative proteins with increased abundance include Carbonic anhydrase 1 and 2 as well as Thioredoxin domain-containing protein 17. Of note, proteomic signature of the muscle biopsy derived from our patient also indicates that loss of functional LDHA impacts on proper cytoskeleton and thus most likely on regular contraction. However, this presumably represents a secondary pathophysiological phenomenon and more functional studies are needed to further elucidate the exact pathophysiological interplay between loss of LDHA, associated metabolic dysfunction accompanied by oxidative stress burden and vulnerability of cytoskeleton. Our proteomic data for instance highlight affected cytoskeleton by decrease of heavy chain myosins 6 and 8 as well as myosin regulatory light polypeptide 9. Activation of the complement system and immune cell activation as unraveled by our immunostaining data accords with the increase of several immunoglobulins and factors of the complement cascade as identified by proteomic profiling. In addition, the increase of several proteins such as XIRP1 (protein aggregation marker) components of the proteasome (PSMA4, PSMD4, UBE2 K & UCHL1) and chaperones (ERP29 & HSPB6) accord with the concept of LDHA deficiency manifesting as a vacuolar myopathy. Hence, our proteomic findings provide significant insights into the pathobiochemistry of the disease.
Since the first family was described in Japan in 1980, 1 only 16 other families have been reported in the literature. Most of the families reported harbour homozygous deletions of 20-bp in exon 6 of the LDHA gene, one splicing mutation and a small insertion and more recently two novel nonsense variants associated with LDHA deficiency were identified in a Spanish cohort.13–18
Footnotes
Acknowledgements including sources of support
The authors acknowledge the financial support of the Association de l’ Institut de Myologie (AIM) et de L’Association Française contre les Myopathies (AFM). AR acknowledges the financial support of the German Society of Muscular Diseases (DGM) as well as of the French Muscular Dystrophy Association (AFM-Téléthon; grant # 21644). Parts of the study were funded by the European Regional Development Fund (project NMD-GPS; ![]() ). Moreover, the support by the “Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen” for MODS [grant no. PROFILNRW-2020–107-A] is acknowledged.
). Moreover, the support by the “Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen” for MODS [grant no. PROFILNRW-2020–107-A] is acknowledged.
AH gratefully acknowledges the financial support by the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen and the Bundesministerium für Bildung und Forschung.
The authors acknowledge the input of Dr François Petit in the genetic discussion of this case.
Several authors ‘are part or the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD).
Funding
The authors received financial support for the research, authorship, and/or publication of this article from the Association de l'Institut de Myologie.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Datasets/data availability statement
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.