
Abstract
PAX7 is a myogenesis transcription factor important for satellite cell specification and function and thus involved in muscle growth, maintenance, repair and regeneration. Recently, a new autosomal recessive congenital myopathy was described that is caused by biallelic variants in PAX7. Our aim is to describe phenotype and whole-body muscle MRI with follow-up imaging findings in a patient with a novel homozygous missense variant in PAX7. We also compare our patients’ imaging features with a patient reported in the initial study, to identify a possible emerging pattern for PAX7-congenital myopathy. Generalized muscle hypotrophy and selective sternocleidomastoid, paraspinal and thigh muscle involvement emerge as suggestive findings and could serve as a recognizable fingerprint to differentiate early-onset myopathies within the emerging group of ‘primary satellite cell-opathies’.
Introduction
The persistent potential of the skeletal muscle to functionally adapt and regenerate is determined by the resident population of muscle stem cells, called ‘satellite cells’, which have a crucial role in growth and homeostasis of skeletal muscle.1–5 Recently, a group of genetic neuromuscular conditions has been delineated predominantly affecting satellite cell function, and defined as ‘primary satellite cell-opathies’.6,7 Neuromuscular disorders, in which the pathogenic mechanism targets both satellite cells and muscle fibres, were defined as the secondary forms.6,7
In 2017, a human autosomal recessive syndrome characterized by severe global developmental delay, failure to thrive, microcephaly, axial hypotonia, pyramidal tract signs, dystonic posture, seizures, irritability and self-mutilation was first described to be due to PAX7 splice-site mutation resulting in nonsense-mediated mRNA decay affecting only PAX7 isoform 3. 8 In 2019, a myopathy caused by biallelic loss of function variants in PAX7 was described in a single report including five individuals from four unrelated families, at the ages of 4–14 years, manifesting with congenital myopathy (CM) characterized by hypotonia, ptosis, mild dysmorphic features, muscular atrophy, and scoliosis. 9 Thereafter, another patient with a similar phenotype at the age of 6 years and a previously identified homozygous c.86-1G > A PAX7 variant was described. 10 This myopathy was referred to as congenital myopathy 19 (CMYO19), progressive CM with scoliosis (MYOSCO; MIM: 618578).9,10 The disease was progressive in nature with weakness in the axial muscles and diaphragm combined with scoliosis that led to respiratory insufficiency. Muscle biopsy samples either had no overt pathological alterations or demonstrated the presence of atrophic fibres and fibroadipose tissue replacement without myofiber necrosis but altered satellite cell numbers.9,10
Here we add to the phenotypic spectrum and definition of PAX7-CMYO by reporting a patient with a novel homozygous missense variant (c.801G > A, p.Asn267Lys) in PAX7 (NM_002584.2) and describe the clinical phenotype as well as whole-body and follow-up magnetic resonance imaging (MRI) findings. Recognition of distinctive patterns of muscle involvement on imaging studies as part of ‘deep phenotyping’ is a helpful tool in the diagnostic pathway of patients with rare neuromuscular diseases.11–15 To fill in this knowledge gap in PAX7-CMYO from an imaging perspective, we further compare the whole-body MRI imaging features of our patient with a previous report 10 using the Mercuri scale to visually grade fat content of individual muscles. 16
Case report
A now 9 years old Turkish girl presented to our pediatric neurology outpatient clinic at the age of 4 years-4 months with a primary complaint of difficulty in walking and abnormal gait. Parents reported a history of decreased intrauterine movements and polyhdramniosis. Pregnancy was otherwise uneventful. She was born at term with normal spontaneous vaginal delivery, and a birth-weight of 1800 g. She was noted to have a weak cry, ptosis, cyanosis and respiratory insufficiency at birth. She stayed at the neonatal intensive care unit for 20 days without requiring intubation. She had swallowing difficulties and a history of recurrent lung infections within the 1st year of life, and required intensive care unit (ICU) admission during the course of a respiratory infection at the age of 8 months. During the follow-up at the ICU, she had a single seizure episode which did not require medication. Thereafter, she has remained seizure free. Early developmental milestones were achieved at normal ages; she achieved head control, sitting without support and independent walking, at the ages of 4 months, 8 months and 14 months, respectively. She was able to climb up a stair with support by the age of 18 months, but has not yet achieved the abiliy to climb stairs without support or jump, to date. Cognitive milestones were normal.
Parents reported that her walk always had a waddling quality, which however had become more prominent in the last three years. Her gait was characterized by inward positioning of the feet with lateral positioning of upper extremities to compensate gait abnormality. Scoliosis was noticed at age two and a half years, which was progressive. By the age of 5 years, she developed signs and symptoms of nocturnal hypoventilation syndrome and required non-invasive bilevel positive airway pressure (BiPAP) thereafter. She always had a low pitched voice. Chewing and swallowing problems have become more prominent at the age of 7 years, and she subsequently developed dysphagia. For the last two years, she was unable to get up from the floor without support and had difficulties with daily functioning including dressing herself and using spoon and fork. Thus, her weakness had progressed affecting both upper and lower extremities with axial and truncal involvement.
Family history degree was significant for consanguinity in that her parents were first cousins. Her 12-year-old brother was healthy. Family history was otherwise unremarkable.
On her recent physical examination, head circumference was 48,5 cm (< 3p); body weight: 20 kg (3–10p), height: 124 cm (25–50p), and body mass index was 13 kg/m2. She had dysmorphic features including bifrontal narrowing, bushy eyelashes, bilateral ptosis, bulbous nose, high arched palate, retrognathia, a myopathic face with hypomimia (Figure 1). Her head was tilted to the right side with limited lateral neck rotations on both sides. She had a scoliosis, inability to flex the knees, knee and foot ankle contractures on the right side, and achilles tendon contractures bilaterally. There was neck flexor (Medical Research Council (MRC): 3/5) and extensor weakness (MRC: 3/5) combined with proximal (MRC: 2+-3/5) and distal (3+/5) upper extremity weakness. She was only able to to move her hands to the shoulder level. Gowers’ sign was positive and she required 9 s for standing up. Lower extremity proximal weakness was prominent at the hip flexors (MRC: 2/5), extensors (MRC: 3/5), knee extension (MRC: 3/5), dorsiflexion (MRC: 3/5) and plantar flexion (MRC: 4/5). Knee flexion was not possible with the right side affected more than the left. She had a waddling, wide-based gait with the upper extremity extended to the back for compensation. She was unable to get up from supine position to sitting position with marked neck and trunk flexor weakness, and head lag (Supplementary Figure 1(a)-(c)).
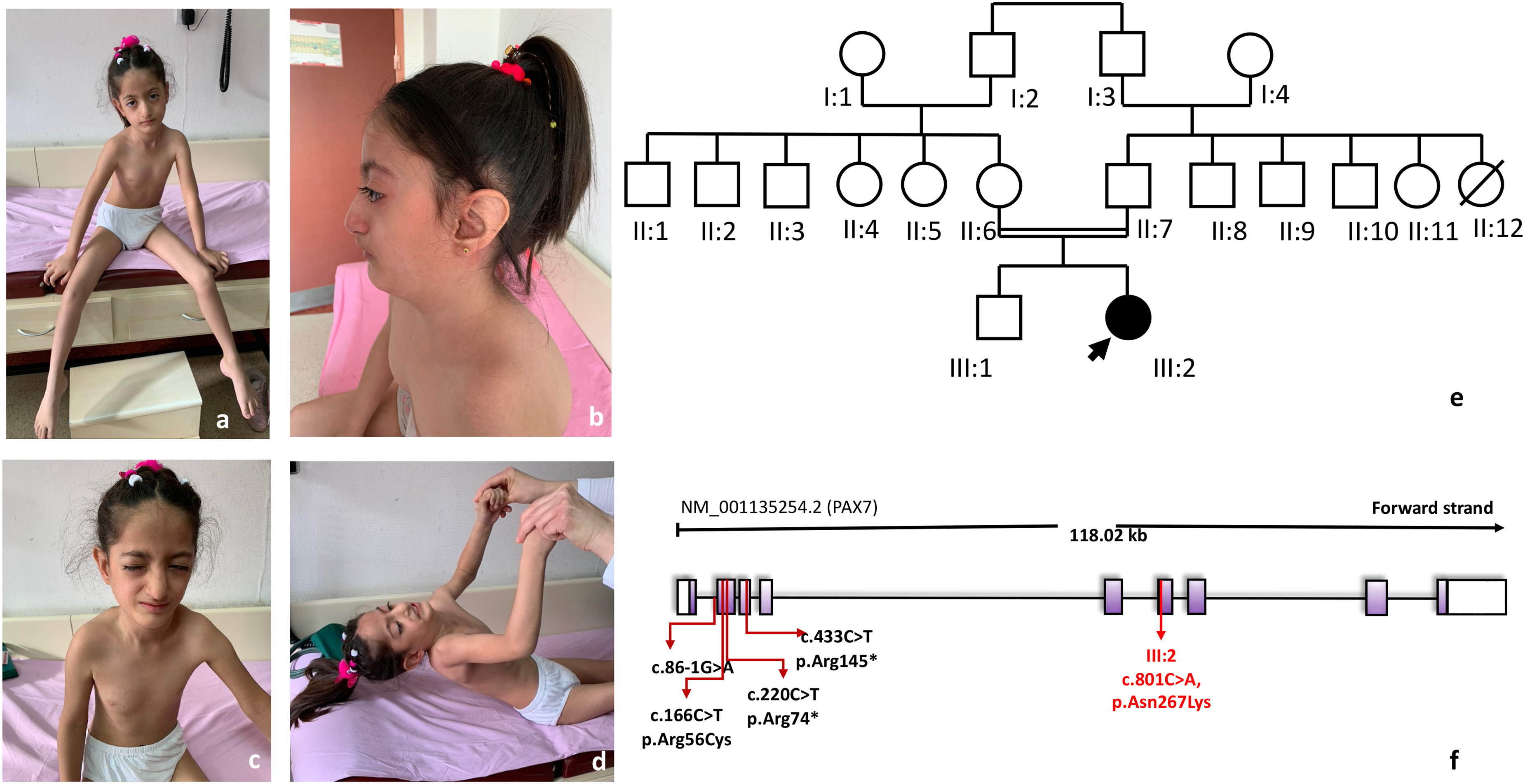
Patient at the age of 8 years: Asthenic phenotype, head tilt, facial dysmorphic features include bilateral ptosis, myopathic face, bifrontal narrowing, bushy eyelashes, bulbous nose, retrognathia, low-set ears (a-c), neck flexor weakness and axial involvement (d). Pedigree (e), and position of the so far identified homozygous variants in the PAX7 gene including this report (f). Written informed consent is obtained for the images presented.
Serum creatine kinase level was normal. Muscle biopsy at the age of 4 years and 4 months from the vastus lateralis muscle showed very mild variation in fibre size with preservation of normal polygonal outline of fibres (Supplementary Figure 2(a)). Immunohistochemical staining with oxidative enzymes (NADH, SDH, COX, COX-SDH) showed uneven staining in a few fibres. SDH and COX stainings were normal. Distribution of type 1 and type 2 fibres was normal on ATPase staining. Spectrin, collagen 6, laminin-211, dystrophin and sarcoglycan stainings were normal. These findings were nonspecific. Spine X-ray demonstrated dextroscoliosis (Cobb angle: 21°C). Fluoroscopic evaluation of diaphragm was unremarkable. Polysomnography study revealed severe hypoxemia during sleep, as well as chronic respiratory insufficiency. Echocardiography revealed mild right chamber dilatation at the age of 4 years and 9 months, which was obtained in the setting of respiratory distress and nocturnal hypoventilation syndrome. Nocturnal non-invasive ventilation using BiPAP was initiated at that time. Forced vital capacity in the upright position was 23% predicted at the age of 6 years. Follow-up echocardiography study at age 8 years was normal.
Whole-body MRI at 6 years and 3 months of age revealed atrophy of bilateral sternocleidomastoid, paraspinal, gluteal, and thigh muscles as well as mild thoracolumbar dextroscoliosis without evidence of vertebral segmentation or spinal cord abnormalities (Figure 2(a)-(d)). Paraspinal muscle involvement comprised erector spinae at lumbar, thoracic and neck levels, and iliopsoas muscles on both sides. One and a half years later, MR images through the thighs and mid-thigh (Figure 2(e), (f)), showed marked and fatty replacement of the adductor (asterisks, Figure 2(e)) and sartorius muscles, and milder and more focal fatty replacement in the quadriceps (the deeper layers of the vastus laterals) and in the semitendinosus muscles in both legs creating a ‘tigroid pattern’ with hypo- and hyperintensity (arrows, Figure 2(f)). Over the interval, there had been an increase in the volume loss and fatty replacement of the gracilis (arrowheads, Figure 2(a), (e)) and sartorius (arrows, Figure 2(a), (e)) muscles. These findings appear fairly symmetric.
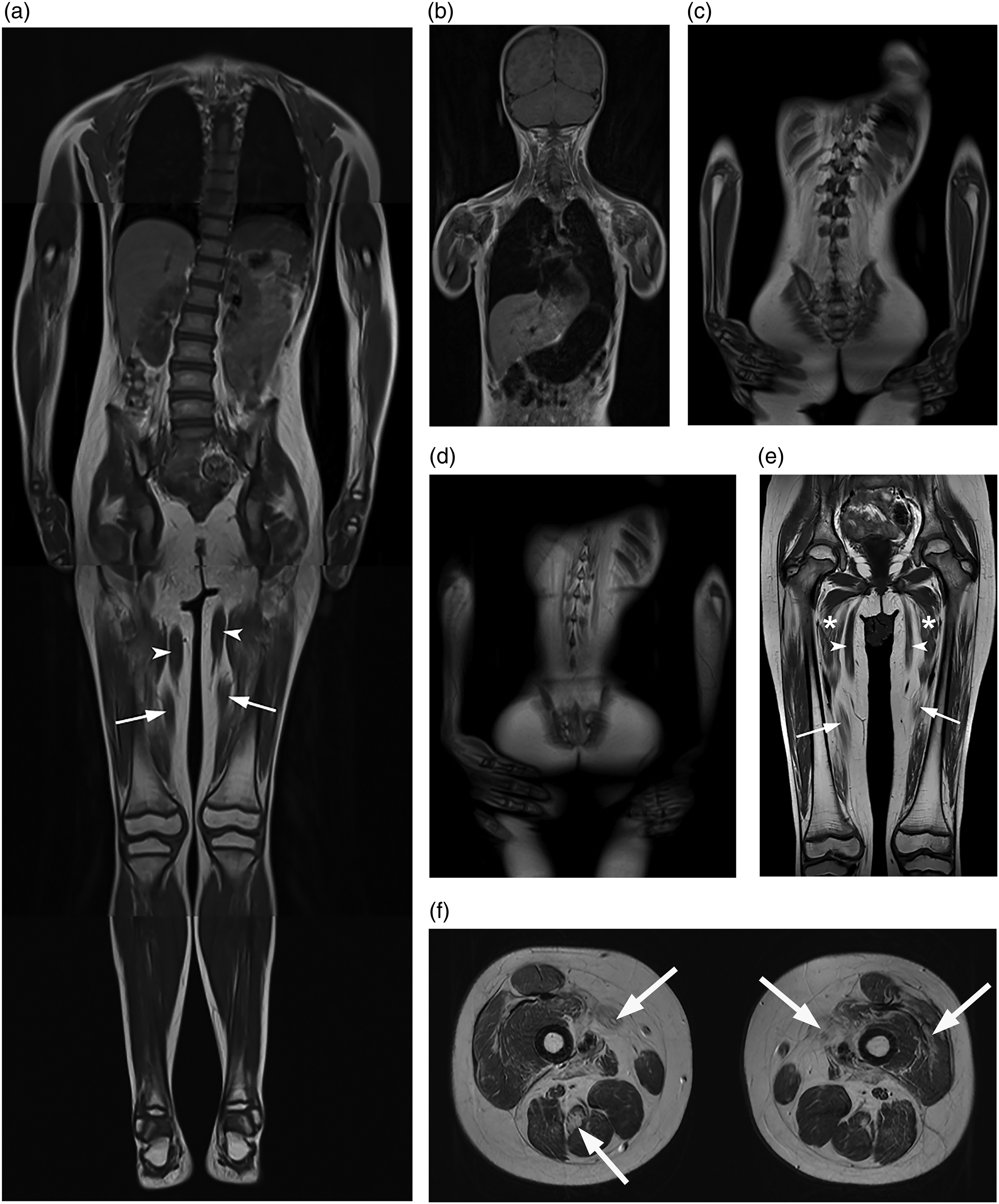
At age 6 years and 3 months, a coronal composite (
Trio exome sequencing identified a homozygous missense variant (c.801G > A, p.Asn267Lys) in PAX7 (NM_002584.2). The c.801G > A, p.Asn267Lys variant is rare, predicted to be damaging (CADD Prediction: 24; MutationTaster: disease causing) and impacts a conserved residue. The variant has been listed once (1/ 206332 alleles) in the heterozygous state in gnomAD v.2.1.1, there are no homozygous individuals in gnomAD. Both parents and the unaffected sibling where heterozygous for the variant, consistent with recessive disease. Pedigree and position of the detected homozygous variants in the PAX7 gene is presented (Figure 1(e), (f)). Re-evaluation of the muscle biopsy revealed absence of PAX7 positive cells (Supplementary Figure 2(b)).
This study is approved by the Hacettepe University Non-interventional Clinical Research Ethics Board and Commission (# 16969557-277), and written informed consent was obtained for images of the patient presented.
Discussion
Here we describe detailed clinical and muscle MRI findings in a patient with a novel homozygous missense variant in PAX7. This patient further expands the clinical and genetic spectrum of a newly emerging recessive myopathy due to pathogenic variants in PAX7, a known master regulator of satellite cell specification and function. Pathogenicity of the variant was supported by muscle biopsy analysis which revealed absence of PAX7 positive cells. We include the initial whole-body and follow-up focused muscle MRI as a complementary tool to aid in the clinical, laboratory, histologic and genetic characterization of this myopathy within the group of ‘primary satellite-cellopathies’. At this time, a single series describing five individuals from four unrelated families with PAX7-CMYO, more recently, another patient with a previously identified homozygous variant has been reported.9,10 All patients presented with hypotonia, dysmorphic features and progressive weakness and scoliosis of variable severity. With our report we confirm the previously noted PAX7-CMYO manifestations. In addition, detailed clinical phenotyping in our patient highlights further findings including predominant axial and truncal involvement with limited neck rotations, head tilt, and dysmorphic features including bifrontal narrowing, bushy eyelashes and a bulbous nose. The disease was preliminary designated as MYOSCO, referring to its overall clinical manifestation of ‘myopathy, scoliosis and congenital onset’.9,10 Keeping in line with recent revisions of the classification systems in neuromuscular diseases, we suggest PAX7-related disease (PAX7-RD), and PAX7-CMYO for the early-onset presentations, as a more unified nomenclature encompassing current and possible emerging future phenotypes. 17
Recognition of differential pattern of muscle involvement as part of ‘deep phenotyping’ supported by MYO-MRI Working Group is becoming increasingly important in inherited myopathies for identification of characteristic patterns of muscle involvement and to aid in genetic variant identification and classification. 11 As we show here, in a clinical setting of patients presenting with early-onset weakness, with normal serum CK levels and relatively nonspecific findings on muscle biopsy, muscle MRI can be a useful non-invasive imaging tool to provide complementary data for the diagnosis of neuromuscular diseases.11–16 Whole-body muscle MRI in our patient revealed atrophy of bilateral sternocleidomastoid, paraspinal, gluteal and thigh muscle involvement at age 6 years. A subsequent muscle MRI through the mid-thigh at age 7 years and 9 months revealed fairly symmetric, marked fatty replacement of the adductor and sartorius muscles, and milder and more focal fatty replacement in the quadriceps (the deeper layers of the vastus laterals) and in the semitendinosus muscles in both legs. The pattern of fatty replacement of the involved muscles can be described as a ‘tigroid pattern’ suggesting involvement of some interspersed muscle fibres along with relative preservation of others. This tigroid pattern defined as alternating bands of hypo- and hyperintensity within multiple muscles is not specific, and can be also observed in limbs and girdles in young patients with neuromuscular disorders and some atypical patients with COL6-RD.12,13 Of interest, in the original report, thigh MRI in a 6-year-old girl with PAX7-CMYO was reported to be normal. 9 It is possible that early in the disease process, muscle MRI may not be sensitive enough to pickup subtle involvement, and muscle ultrasound may be a more appropriate complementary modality. 18
Comparison of whole-body MRI with the previously published patient 10 showed an emerging pattern of muscle involvement (Supplementary Figure 3(a)-(c)). Graphical representation of the Mercuri scores (Figure 3) reflect a shared imaging pattern characterized by a general muscle hypotrophy with involvement of occipital, sternocleidomastoid, paraspinal and thigh muscle, and sparing of distal muscles. Of interest, there is a selective and partial involvement of semitendinosus muscle in both of these patients. Moreover, a follow-up image of our patient at the mid-thigh level demonstrates progressive atrophy and fatty infiltration of the adductors and sartorius muscles with milder and focal involvement of quadriceps.
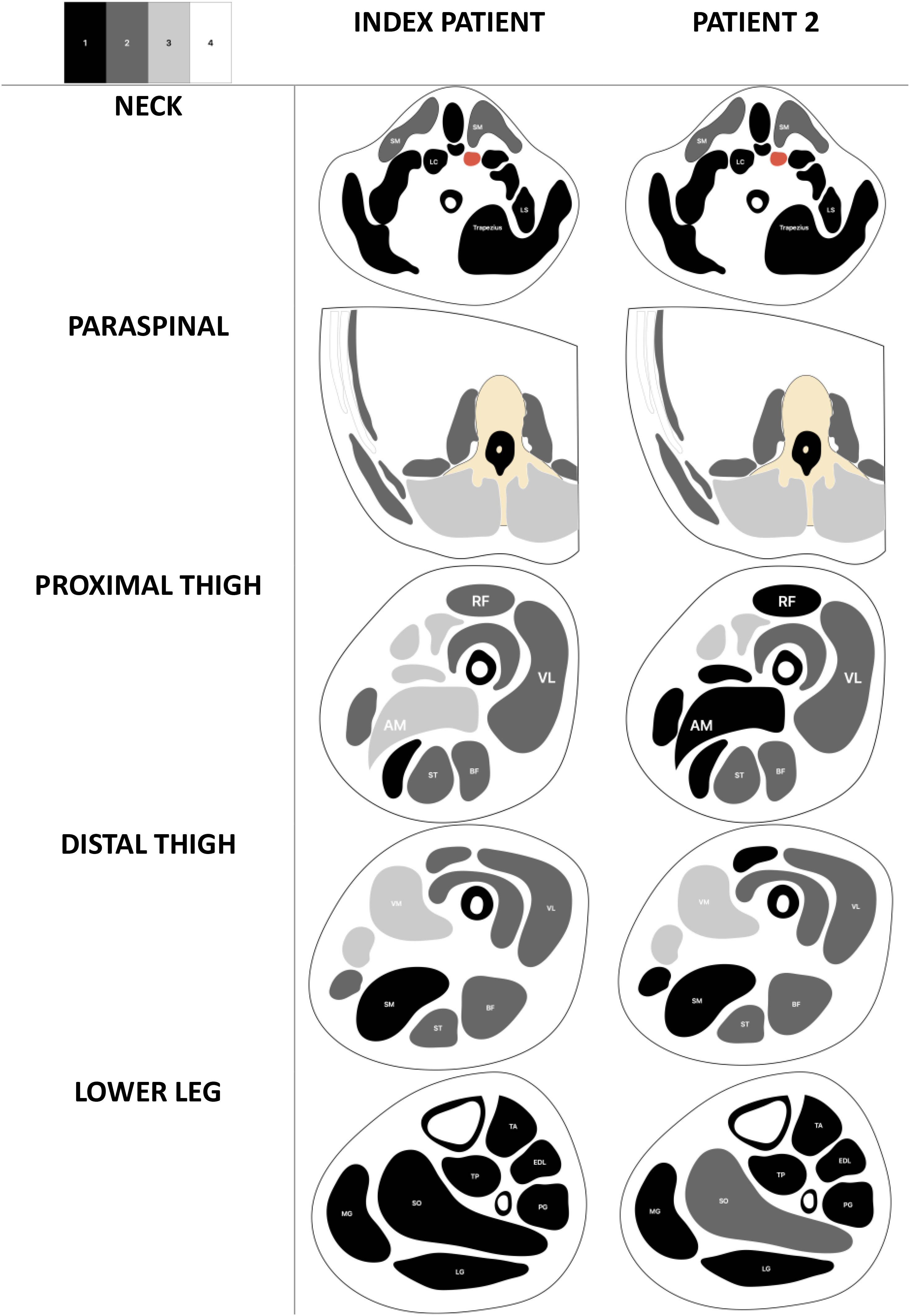
Graphical representation of the whole-body MRI images in PAX7-CM. Cross-sectional view through neck, paraspinal, upper, mid and lower-thigh levels of the patient at age 6 years-3 months (Patient 1) in comparison with the previously reported patient at age 6 years. 10
Among the myopathies, pathogenic variants in SELENON, TTN, NEB, TPM3, ACTA1, LMNA, and COL6A1-3 may also present with axial predominant, progressive weakness, scoliosis, and respiratory involvement. 19 Identification of hallmark patterns of muscle involvement in this sub-set of myopathies continue to emerge, to now also include PAX7-CMYO. Hankiewicz et al. described a homogeneous pattern of involvement by whole-body MRI in SELENON-related myopathies including severe atrophy of sternocleidomastoid muscle, selective paraspinal, gluteus maximus and thigh muscle involvement with atrophy and marked signal abnormality of semimembranosus and also sartorius. 20 Differential diagnosis of axial involvement and sternocleidomastoid atrophy also include DNM2 variants.11,12 Paraspinal, gluteal and hamstring atrophy with sparing of adductors, sartorius and gracilis were reported in TTN-related myopathies along with selective involvement of semitendinosus. 21
The progression of weakness seen in patients with biallelic variants in PAX7-CMYO is thought to be related to an exhaustion of the satellite cell pool, which overtime results in impaired muscle growth and regeneration capacity.1–7 In addition, we see progression on muscle imaging with an increase in fatty replacement of muscle over time as shown in our patient's muscle MRI imaging. This may reflect the decrease in the stem cell pool and self-renewal capacity of the skeletal muscle to functionally adapt and regenerate to any stimuli, such as growth cues, exercise, trauma, injury, etc.
The congenital findings of ptosis, dysmorphic features, weak cry and respiratory features noted at birth in our patient do point towards an additional developmental role for PAX7. 22 Mouse models have provided important insights in the expression pattern of Pax7 highlighting its role in the development of cephalic neural crest derivatives, the central nervous system, and skeletal muscle.22,23 Pax7 null mice are not viable beyond 3 weeks of life, display malformations in facial structures including maxilla and nose, with surviving mice being significantly small in size and weight.22,23 Diaphragmatic involvement, respiratory insufficiency, dysphagia, dysmorphic facial features are common features shared by PAX7-CMYO and other neuromuscular disorders where variants disrupt satellite cell myogenesis and self-renewal, including MYOMAKER (MYMK) causing Carey-Fineman-Ziter syndrome, and SELENON, MEGF10, MYOD, MYMK and JAG2. 7
In summary, in this report we describe the presentation and deep phenotype including pattern of muscle involvement by whole-body as well as focused follow-up MRI findings in a patient with a novel homozygous p.Asn267Lys missense variant in PAX7, a novel case that adds to the emerging primary satellite cell-opathies. Our findings confirm the previously described clinical features, in particular childhood onset progressive weakness, scoliosis, respiratory insufficiency and facial dysmorphisms in the setting of normal serum CK levels, to now also include detailed muscle MRI findings and a recognizable imaging pattern.
Targeted gene panels for early-onset muscle diseases (CMDs and CMs) should include PAX7. Taking into consideration that three of the reported six patients so far had either pyridostigmine trial or repetitive nerve stimulation tests, undiagnosed congenital myasthenic syndromes can be another target. Although not completely specific, availability of antibodies against PAX7, NCAM, M-Cadherin and CD56 can be used and facilitate the diagnosis of satellite cell-opathies through direct gene testing. 7 Clinical characterization of additional patients with pathogenic PAX7 variants will be helpful to further expand the natural history and clinical, histopathological and imaging spectrum of this rare myopathy.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602241289705 - Supplemental material for Distinct whole-body muscle MRI imaging patterns in PAX7-congenital myopathy: A case report
Supplemental material, sj-docx-1-jnd-10.1177_22143602241289705 for Distinct whole-body muscle MRI imaging patterns in PAX7-congenital myopathy: A case report by Göknur Haliloğlu, Sandra Donkervoort, Sibel öz Yıldız, Abigail Potticary, Ying Hu, Lynn Pais, Robert Y. Carlier, Helge Amthor, Üstün Aydıngöz and Carsten G. Bönnemann in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
We thank our patient and her family for their participation. Authors thank Can Koşukcu from Hacettepe University Departments of Bioinformatics, Dr Neşe Vardar from Hacettepe University Molecular Biology for Figure 1(f), and Dr Beril Talim from Hacettepe University Faculty of Medicine, Department of Pediatrics, Pediatric Pathology Unit for Supplementary ![]() . C.G.B.'s laboratory is supported by intramural funds from the NIH National Institute of Neurological Disorders and Stroke. G.H. is supported by GREGoR Consortium, and research in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG011746.
. C.G.B.'s laboratory is supported by intramural funds from the NIH National Institute of Neurological Disorders and Stroke. G.H. is supported by GREGoR Consortium, and research in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG011746.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Ü.A. received speaker honoraria from GE HealthCare. Other authors have no conflict of interest to report. C.B.G is Editor-in-Chief and G.H. is an Editorial Board member of this journal but were not involved in the peer-review process nor had access to any information regarding its peer review.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.