
Abstract
The resident stem cell for skeletal muscle is the satellite cell. On the 50th anniversary of its discovery in 1961, we described the history of skeletal muscle research and the seminal findings made during the first 20 years in the life of the satellite cell (Scharner and Zammit 2011, doi: 10.1186/2044-5040-1-28). These studies established the satellite cell as the source of myoblasts for growth and regeneration of skeletal muscle. Now on the 60th anniversary, we highlight breakthroughs in the second phase of satellite cell research from 1980 to 2000. These include technical innovations such as isolation of primary satellite cells and viable muscle fibres complete with satellite cells in their niche, together with generation of many useful reagents including genetically modified organisms and antibodies still in use today. New methodologies were combined with description of endogenous satellite cells markers, notably Pax7. Discovery of the muscle regulatory factors Myf5, MyoD, myogenin, and MRF4 in the late 1980s revolutionized understanding of the control of both developmental and regerenative myogenesis. Emergence of genetic lineage markers facilitated identification of satellite cells in situ, and also empowered transplantation studies to examine satellite cell function. Finally, satellite cell heterogeneity and the supportive role of non-satellite cell types in muscle regeneration were described. These major advances in methodology and in understanding satellite cell biology provided further foundations for the dramatic escalation of work on muscle stem cells in the 21st century.
Keywords
INTRODUCTION
Skeletal muscle is composed of syncytial muscle fibres in which many myonuclei control a common cytoplasm containing the sarcomeres that generate force by contraction. Muscle fibres are formed by fusion of mononuclear myoblasts into myotubes, which then grow by addition of further myonuclei from myoblast fusion and hypertrophy. Myonuclei are post mitotic, so repair and regeneration of skeletal muscle fibres requires a source of myoblasts to either fuse to existing muscle fibres to donate new myonuclei, or fuse together for de novo myofibre formation. The muscle satellite cell is accepted as the predominant resident stem cell source supplying myoblasts to postnatal skeletal muscle [1, 2].
On the 50th anniversary of the discovery of the satellite cell by Alexander Mauro [3] (and Bernard Katz on intrafusal myofibres [4]) in 1961, we first discussed the early studies in skeletal muscle growth and regeneration, starting in the 19th century when most publications were in scholarly German. We then considered the key findings up to ∼1980, by which time the satellite cell had generally become accepted as the preeminent source of myoblasts for growth and regeneration of skeletal muscle [5].
In this companion review in the 60th year of the satellite cell, we focus on the second phase of satellite cell research from 1980 to 2000, which includes some of the major discoveries that laid further foundations for satellite cell research. We start by highlighting the technical innovations for purification of primary myogenic cells [6], and isolation of viable muscle fibres complete with satellite cells in their niche [7, 8]. This is followed by discussion of endogenous markers that identify quiescent satellite cells in situ at the light microscopic level, the most important being the transcription factor Pax7 [9] (especially since modification of the Pax7 locus allows specific targeting of satellite cells [1]). The seminal discovery of the muscle regulatory factor (MRF) family of transcription factors helped explain muscle formation and linked control of developmental and regenerative myogenesis [10], as well as provide important tools such as excellent antibodies to MyoD [11] and myogenin [12]. Genetic modification to explore the regulation, expression and role of myogenic genes further enhanced understanding of satellite cell function [10]. Genetic engineering also delivered early lineage markers such as the Myf5 nlacZ allele [13] and 3F-nlacZ-E transgene [14], which facilitated identification of satellite cells in situ [15]. Importantly, such lineage markers also allowed cell fate to be discerned following transplantation, permitting the full potential of grafting experiments to be realized. This ultimately led to the designation of satellite cells as stem cells in 2005 [16], with subsequent studies confirming this classification [17, 18]. Finally, early work on satellite cell heterogeneity and the support of muscle regeneration by non-satellite cells are discussed, along with initiation of the Satellite Cell Conference series in 1998. Important in themselves, studies in this second phase of satellite cell research also underpinned the surge of investigation in muscle stem cells in the 21st century [19].
METHODS TO OBTAIN SATELLITE CELLS
Generation of clonal myogenic cell lines allowed the study of myogenesis to become more accessible, but many of these early lines were derived from developing muscle [5]. A number of clonal myogenic lines derived from adult mouse muscle were developed in the late 1970s/early 1980s that were more likely to be satellite cell in origin. Of these, the C2 cell line is probably the best known, derived by Yaffe and Saxel in 1977 [20], which was later subcloned to generate the C2C12 cell line [21], which is still widely used. Other notable satellite cell-derived mouse lines include MM14 and its derivatives [22, 23] and H-2Kb-tsA58 myoblast clones isolated from transgenic mice carrying a thermolabile T antigen [24]. Conditional immortalization now allows routine creation of myoblast clones, including from human e.g. [25]. Those isolated from patients provide versatile disease models, with a good example being the isogenic clones with/without the pathogenic mutation derived from a mosaic FSHD patient [26].
Collecting a representative population of primary satellite cells from adult tissue was more problematic however, and much early investigation was performed using embryonic/foetal myoblasts, for example into the role of growth factors in myogenesis (for contemporary review see [27]). While trypsin, pronase, collagenase, papain or ficin could be used to liberate myoblasts from embryonic chick and foetal rat muscle, Richard Bischoff found that only trypsin and pronase proved effective in adult rat [28, 29]. Thus adult muscle needed proteases to release satellite cells from the basal lamina of the myofibre. Additionally, although a high proportion of myogenic cells were obtained, fibroblasts would often overgrow the cultures [28, 29]. In 1981, Blau and Webster optimised pre-plating to enrich populations of myoblasts from adult human muscle tissue [30] and from patients with various diseases [31]. Fibroblastic cells more readily adhere to non-coated cell culture plates so could be removed, allowing the less-adherent myogenic cells to be concentrated and collected. Pre-plating remains part of some protocols e.g. [32].
A drawback of pre-plating enrichment strategies was that cell debris and myofibre fragments remained in the cell suspension, preventing analysis immediately following isolation. Combination of pronase digestion, differential centrifugation and pre-plating improved yield, permitting Ron Allen and colleagues to show that fibroblast growth factor (FGF) promoted rat satellite cell proliferation [33, 34]. Percoll fractionation, where myogenic cells occupy a specific layer following density centrifugation, removed additional debris, producing an enriched myogenic population [35, 36], although not all contaminating cells were removed [37]. However, these methods had an important caveat, that there was “no way of clearly identifying the myogenic precursor cells isolated from the adult muscle as the ‘classical’ satellite cells” [35].
Advent of fluorescence-activated cell sorting (FACS) was a game changing technology for purifying primary myogenic cells from adult muscle. Antibodies that bound satellite cells (e.g. against N-CAM) allowed a relatively pure population of myogenic cells to be obtained from adult muscle [6]. FACS remains a standard to isolate primary satellite cells, using a combination of endogenous antigens for positive and negative selection [38], and/or genetic modifications such as Pax7-ZsGreen [39]. A drawback of FACS is that it is somewhat technical and does require expensive equipment and skilled support, but bench-top methods such as magnetic bead cell sorting are also effective e.g. [40]. These techniques facilitated examination of a more representative group of primary myoblasts, rather than the greatly expanded progeny of a few select clones.
ISOLATION OF SATELLITE CELLS IN THEIR NICHE ON A MYOFIBRE
To better investigate satellite cells and muscle regeneration, early studies of Irwin Konigsberg et al. [41, 42] and Richard Bischoff [43] had physically peeled myofibres from muscle fragments “much the same way that a piece of adhesive tape is removed from a surface” [43]. Juvenile quail, human [41, 42] and adult rat muscle [43] all yielded muscle fibres that could then be cultured to produce myogenic cells capable of differentiating into myotubes. Pertinently though, it was noted that while myogenic cells emigrated from some myofibres of younger animals, cells only emerged from the cut ends of adult myofibres, or if the basal lamina was deliberately damaged [29]. While such physical separation from adult muscle left the basal lamina intact, it often damaged the myofibre [29]. This provided a useful tool to study myofibre repair since the satellite cells were unable to traverse the basal lamina and so were retained near the muscle fibre [43], giving a relatively faithful model of events observed later by in vivo imaging of muscle repair [44].
Kopriwa and Moss had shown that muscle fibres could be liberated from growing rats with collagenase, using collagenase digestion of fixed strips of tibialis anterior to identify cells that had been dividing in vivo [45]. Collagenase and protease digestion of unfixed frog cutaneous pectoris by Betz and Sakmann isolated single muscle fibres with satellite cells that were beginning to detach [46]. Bekoff and Betz then employed digestion of unfixed rat flexor digitorum brevis (FDB) muscle with collagenase, followed by mechanical trituration to separate live myofibres, before plating the suspension in collagen-coated dishes [47] (Fig. 1A). However, the majority of myofibres remained in suspension where they formed clumps, meaning that only the small number of muscle fibres that adhered to the dish could be analysed. Primarily interested in using myofibres to investigate the effects of denervation such as acetylcholine sensitivity, conditions were not optimized for satellite cells. Dissociated myofibres were not separated from other cell types, and it was noted that the few adherent muscle fibres appeared to be attached by mononuclear cells, which they suspected were fibroblasts [47].
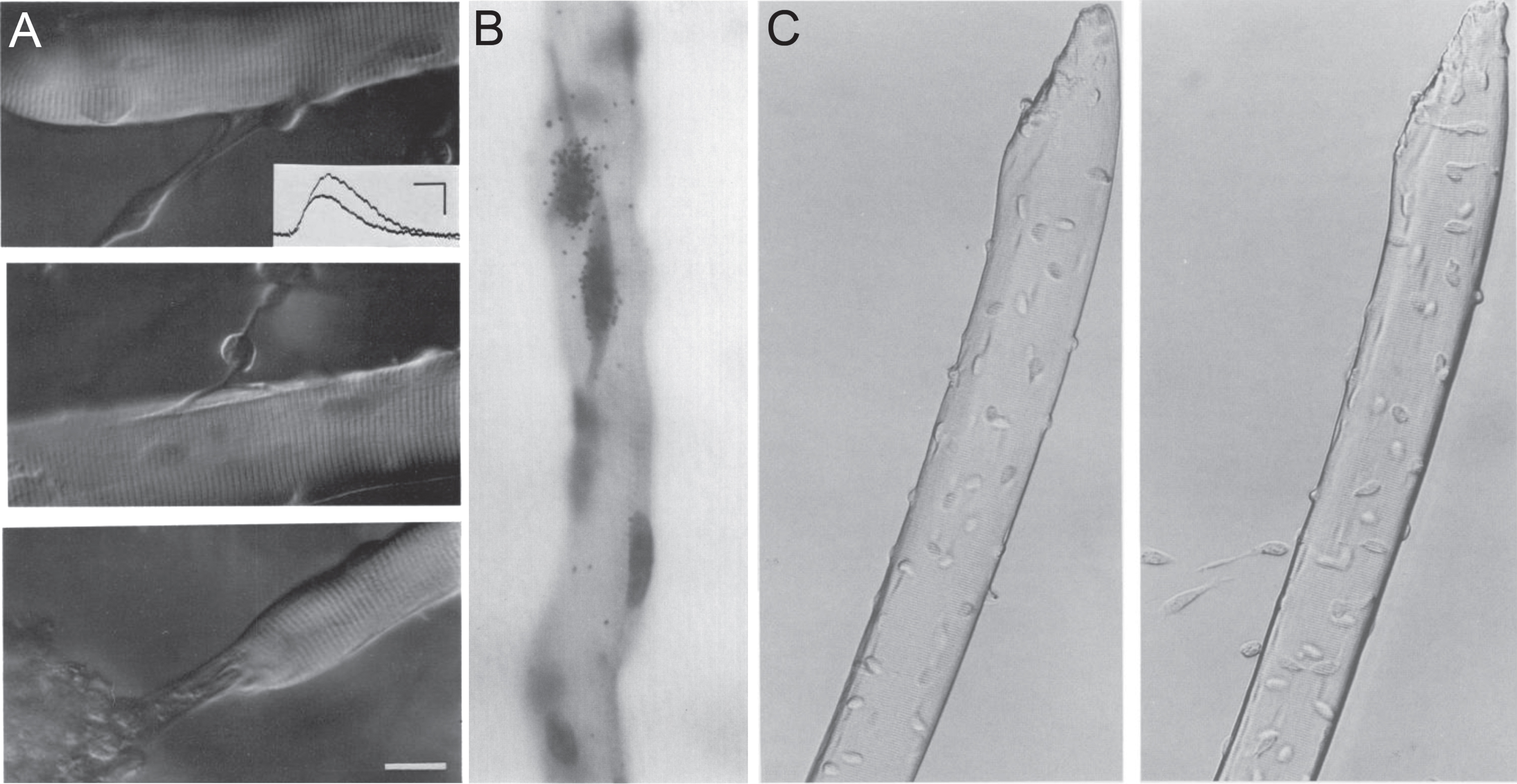
Isolation of muscle fibres to obtain satellite cells in their niche. (A) Three examples of muscle fibres isolated from the flexor digitorum brevis of adult rat and cultured ex vivo for 6-9 days, exhibiting attached cellular processes, from Bekoff and Betz, 1977 [47]. The authors were interested in modeling denervation, with this figure informing on acetylcholine sensitivity of isolated fibres using an intracellular electrode (inset). Scale bar represents 20μm. (B) Muscle fibre from the flexor digitorum brevis of an adult rat from Bischoff, 1986 [8], isolated using a protocol optimized from Bekoff and Betz [47] to better preserve the satellite cell niche. Bischoff was investigating proliferation of satellite cells on isolated myofibres ex vivo and the effects of mitogens. Proliferation was measured via incorporation of tritriated thymidine and autoradiography (generating black grains of silver over radioactive areas), with Gill’s haematoxylin used to identify nuclei. The strongest effect on proliferation was from addition of chicken embryo extract (pictured here), followed by FGF. Magnification is x660. (C) Plated isolated myofibres from the extensor digitorum longus of an adult mouse with migrating satellite cells after 2 hours (left) and 24 hours (right) ex vivo, from Rosenblatt et al., 1995 [7]. The protocol was modified to allow isolation of longer myofibres from larger rodent muscles. Over time, satellite cells detach from the surface of the myofibre and proliferate on the matrigel substrate. Magnification is x300. Images as originally published but may have been cropped, and panel labels have been amended/rearranged/added.
With more focus on satellite cell biology, Rubin et al. reported that a layer of irradiated fibroblasts improved the adherence of rat FDB muscle fibres, which then allowed satellite cells to form myotubes on the culture substrate [48]. Nomarski (differential) interference contrast microscopy was used by Lawrence and Mauro to observe living satellite cells at the light microscope level on a single layer of myofibres from the frog cutaneous pectoris [49]. Application of Nomarski interference to isolated myofibres allowed Cull-Candy et al. to describe live frog satellite cells as fusiform with long fine processes, and typically orientated parallel to the axis of the muscle fibre [50]. Crucially, this approach permitted satellite cell distribution and number to be determined, revealing that satellite cells were arranged with neither pattern nor at consistent density along a muscle fibre, with 4–14 per lumbricalis myofibre.
Bischoff built upon the method of Bekoff and Betz [47] to develop a technique to specifically retain satellite cells on muscle fibres ex vivo [8]. In his modified protocol, Bischoff removed the trypsin-like peptidase clostripain from the collagenase digestion medium, noting that the basal lamina then remained intact using immunolabeling for laminin. A sedimentation step prior to plating was also introduced that purified fibres from tissue debris and mononuclear cells. Difficulty in separating longer muscle fibres confined his studies to muscles containing shorter myofibres such as the FDB of the rat. This protocol meant that satellite cells remained viable for several days ex vivo in their native niche on a myofibre that had adhered to the collagen substrate, allowing recovery of satellite cells from an altered, rounded morphology immediately following isolation. Bischoff investigated factors influencing satellite cell proliferation, as assessed by incorporation of tritiated thymidine and autoradiography. This revealed that chicken embryo extract (CEE) or extract from crushed adult muscle stimulated satellite cell proliferation [8, 51] (Fig. 1B).
Yablonka-Reuveni and Rivera 1994 then expanded the potential of the isolated muscle fibre model by immunolabelling the actual satellite cells in situ on rat FDB myofibres, monitoring changes occurring over several days ex vivo [52]. Using a panel of antibodies, they described the temporal sequence of satellite cell activation, proliferation and differentiation (see below), and also noted how FGF stimulated satellite cell proliferation [52, 53].
In 1995, the Partridge group made further improvements to produce a definitive method to isolate myofibres from larger rodent muscles (extensor digitorum longus (EDL), soleus and tibialis anterior), and to then separate satellite cells/myogenic precursors from the myofibres [7]. While proteases had been used previously to digest the basal lamina to liberate myogenic cells [28], a major modification included defining an optimum range of protease content in the collagenase used to digest the muscle. Other innovations included cleaning by serially transferring to fresh medium, and gentle trituration using a series of heat-polished pipettes with decreasing apertures, which facilitated survival of the longer muscle fibres that were usually lost/damaged by a sedimentation step. Repeating digestion after removal of the exterior-most myofibres allowed isolation of interior myofibres from bulkier muscles. Another tweak was use of Matrigel, extracted from Engelbreth-Holm-Swarm mouse sarcomas [54, 55]. Matrigel is a solubilized preparation rich in extracellular membrane factors including laminin, nidogen, collagen IV and heparan sulfate proteoglycans, available with, or depleted of, growth factors. Matrigel had been shown to be an effective substrate for myogenesis in plated cells [56] but also facilitated attachment of isolated muscle fibres. Crucially, the muscle fibres could also be easily removed, and analysed if desired [7].
With this protocol, Rosenblatt et al. observed satellite cells detaching from adult myofibres within 24 hours of plating and adhering to the Matrigel-coated plate (Fig. 1C), indicating sufficient digestion of the basal lamina. Immunolabeling for the muscle-specific intermediate filament desmin [57] revealed the purity of the satellite cell-derived myoblasts surrounding a myofibre. Collection of the muscle fibres then allowed determination of the muscle fibre type via Western blot and facilitated further myoblast expansion and formation of sizeable myotubes [58]. Alternatively, fixation of myofibres immediately after digestion and trituration allowed examination of near-quiescent satellite cells in their niche [15], since the isolation procedure can initiate satellite cell activation [59, 60].
Isolating satellite cells and muscle fibres provided new opportunities to investigate factors that drive satellite cell activation, including Hepatocyte Growth Factor (HGF) [61, 62], nitric oxide (NO) [63] and mechanical stimulation [64], and proliferation [8, 52]. These techniques also facilitated exploration of differences between satellite cell populations from different fibre types [58], ages [65], dystrophic models [66] and genetically modified backgrounds [67]. Comprehensive protocols for isolation of myofibres and immunolabeling are available [59, 68–70].
SATELLITE CELL MARKERS
For many years following their discovery using electron microscopy, quiescent satellite cells were identified by their anatomical location between the plasmalemma of a muscle fibre and the ensheathing basal lamina. This anatomical niche was used to identify satellite cells on growing myotubes as the basal lamina forms during embryonic day (E) 18/19 of mouse development [71, 72]. However, it was found that satellite cells were likely specified before they could be distinguished on this anatomical criterion in late foetal development.
Embryonic and foetal myoblasts were known to be distinct myogenic lineages (e.g. [73]). Later, differential response of myoblast populations to a tumour promoter indicated that satellite cells also composed a separate lineage, emerging around E16/17 [74], with other characteristics such as acetylcholinesterase isoform [75] and myosin heavy chain (MyHC) content [76] after differentiation providing further support. In chicken, differences in myoblast length, initiation of DNA synthesis and MyHC isoform expression also revealed that adult satellite cells/myoblasts appeared in mid-foetal development [76–78].
The necessity to identify satellite cells on an ultrastructural criterion limited study, especially of quiescence and early activation. While general markers of cell proliferation such as PCNA [79] or incorporation of tritiated thymidine [80] could be used to identify proliferating satellite cell-derived myoblasts in mature tissues, these markers were also expressed in other proliferating cell types. Desmin was observed in the cytoplasm of proliferating satellite cells [37] and in quiescent satellite cells [81], but expression in myofibres limited its use on muscle sections [81] and isolated myofibres.
Indirect identification of satellite cells at the light microscopic level became possible when Zhang and McLennan used antibody labeling of the myofibre plasmalemma with dystrophin and the surrounding basal lamina with collagen IV to distinguish satellite cells from myonuclei [81] (Fig. 2A). Interestingly, it was also noted that dystrophin was present on some satellite cells [81], as later explored in detail [82].
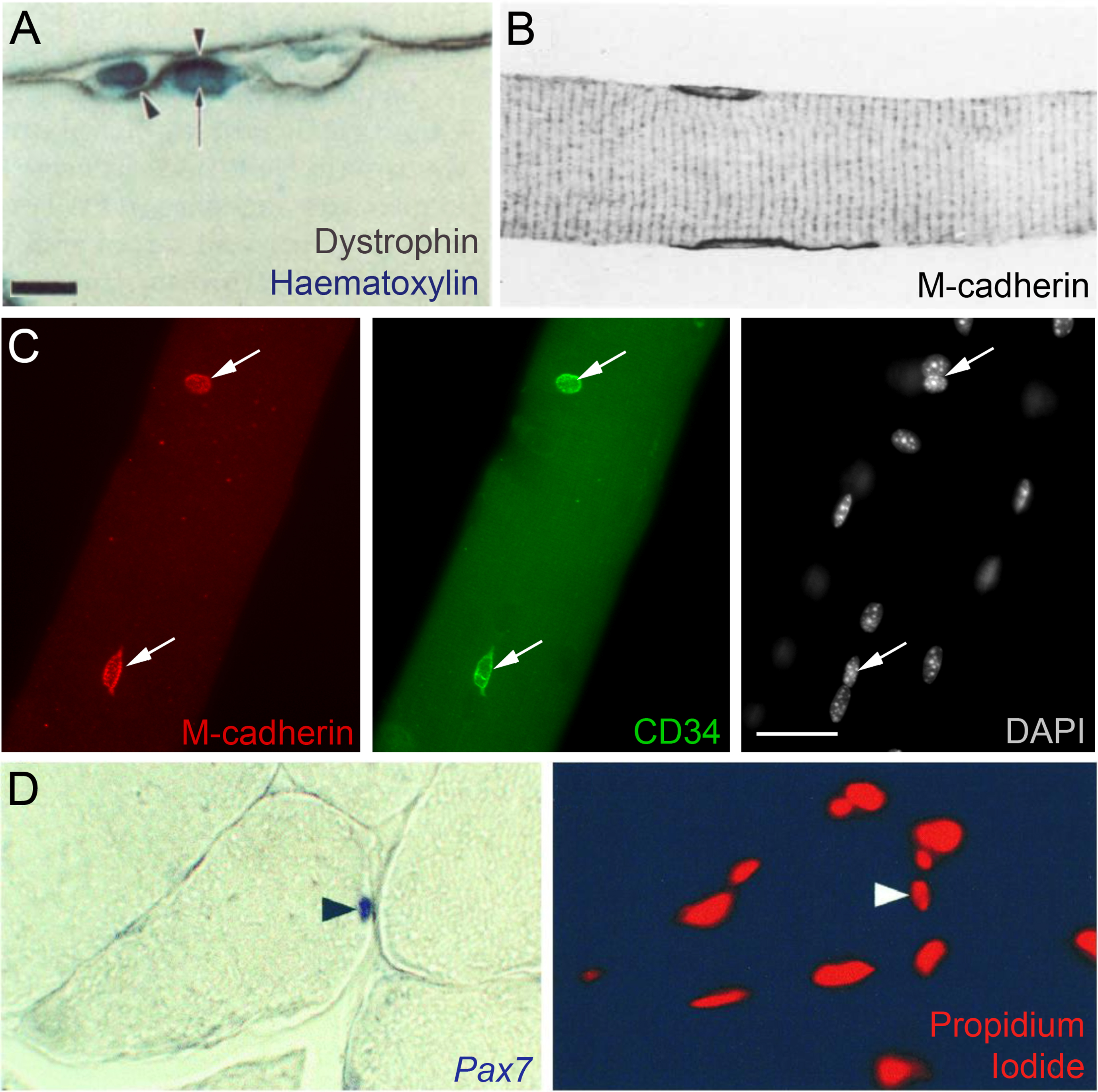
Markers to identify satellite cells in situ. (A) Indirect identification of a satellite cell in a rat muscle using immunolabeling from Zhang and McLennan, 1991 [81]. Dystrophin marks the plasmalemma of the muscle fibre (arrowheads) and two nuclei are visualized using haematoxylin. Satellite cells were distinguished from other nuclei including myonuclei (arrow) by their location outside the dystrophin-labeled plasmalemma but under the basal lamina (using collagen IV immunolabelling - not shown). Scale bar equals 10μm. (B) M-cadherin was shown to mark satellite cells in two papers in 1994 [90, 91], illustrated here with an image of ‘normal’ rat soleus muscle from Bornemann and Schmalbruch, 1994 [91]. The longitudinal section shows two satellite cells labeled with M-cadherin, which is mainly located at the satellite cell/muscle fibre interface. Magnification is x660. (C) CD34 on two satellite cells on an isolated mouse extensor digitorum longus myofibre, confirmed as satellite cells by M-cadherin co-immunolabeling (arrows), and distinct from myonuclei as revealed by DAPI nuclear counterstaining, from Beauchamp et al., 2000 [15]. Scale bar represents 30μm. (D) Pax7 as a satellite cell marker from Seale et al., 2000 [9]. In situ hybridization was used to show that Pax7 mRNA was expressed in approximately 5% of peripherally-located muscle nuclei in healthy adult mouse tibialis anterior muscles (left) with a propidium iodide nuclear counterstain (right). Arrowheads indicate the Pax7-expressing nucleus. Magnification is x200. Images as originally published but may have been cropped, and the panel labels have been amended/rearranged/added.
Direct identification of satellite cells became possible using a monoclonal antibody against leukocyte antigen Leu-19 to visualize satellite cells by Starzinski-Powitz and colleagues [83]. Around the same time, it was revealed that Leu-19 was CD56 or neural cell adhesion molecule (N-CAM) [84], a membrane glycoprotein expressed in neural tissue and on cells surrounding the neuromuscular junction [85]. Expression of N-CAM in healthy extra-junctional adult muscle was confined to a few cells on the periphery of myofibres. Leu-19/N-CAM immunolabelling of satellite cells and myoblasts became a commonly used method for satellite cell identification and, being a surface antigen, was used for FACS-mediated isolation of satellite cells [6]. Increased numbers of N-CAM expressing cells were noted in muscle biopsies of patients with myopathic diseases [86] and mouse models of muscular dystrophy [87].
The next important satellite cell marker was M-cadherin, a muscle-specific member of the cadherin family of calcium-dependent cell-cell adhesion factors. M-cadherin was originally identified in differentiating mouse C2 myoblasts by Starzinski-Powitz and colleagues and was also expressed by myoblasts during development [88]. M-cadherin was linked to satellite cells when a transient wave of M-cadherin mRNA was detected in activated satellite cells following injury [89]. Two independent groups built on this observation, publishing histological studies demonstrating that M-cadherin was on the surface of rodent quiescent satellite cells in vivo [90, 91] (Fig. 2B) but not on other cell types in uninjured muscle [90]. M-cadherin was also expressed in activated and proliferating satellite cells [89, 90]. Unfortunately, there was no effective commercial antibody available, which limited its use.
HGF was found to induce activation of freshly isolated rat satellite cells by Allen and colleagues [61], which indicated that the HGF receptor C-Met should be expressed by quiescent satellite cells. C-Met was detected on the surface of both quiescent and activated satellite cells [61, 92], with low levels also on newly formed myotubes [93] and connective tissue cells [94]. While part of an important signaling pathway for satellite cell activation and a useful candidate marker for quiescent satellite cells [53, 62], antibodies against C-Met available at the time were of variable effectiveness.
Myocyte nuclear factor (MNF, or Foxk1) was also introduced as a satellite cell marker during this period. While MNF was transiently expressed in several non-muscle cell types during development, MNF protein was restricted to the nuclei of quiescent satellite cells in adult muscle, as well as in proliferating myoblasts and the centrally located nuclei of regenerating fibres [95]. Isoforms MNF-α and MNF-β were reciprocally expressed in satellite cell progeny versus quiescent satellite cells, offering the advantage of distinguishing quiescent from activated satellite cells [96].
Screening for cell surface markers known to characterise other stem cell populations, such as those in the haematopoietic system, led to our identification of CD34 as a marker of satellite cells (Fig. 2C), and demonstrated a switch in isoform upon activation [15]. A commercially available biotin-conjugated antibody revealed CD34 on quiescent satellite cells on isolated myofibres, and could also be used as part of a panel of antibodies for FACS. However, expression in other cell types (e.g. endothelial cells) made CD34 difficult to use in muscle sections [97] and it is not present on human satellite cells [98].
The turn of the century saw introduction of Paired box 7 (Pax7) as a satellite cell marker by Michael Rudnicki and colleagues [9]. Pax7, together with the closely related Pax3, were shown to be expressed during both developmental neurogenesis and myogenesis [99, 100]. Pax7 was originally identified in the satellite cell lineage through a series of cDNA subtraction hybridization studies [101]. In situ hybridization showed that Pax7 was expressed in both quiescent satellite cells (Fig. 1D) and proliferating myoblasts, before being down regulated upon myogenic differentiation [9]. At the protein level, the Fujisawa group had reported that Pax7 was present in the myotome and the nuclei of migrating myoblasts forming the trunk and limb muscle precursors in chick embryos, using their own monoclonal antibody [102]. Pax7 immunolabelling was mentioned in Seale et al. [9], with satellite cells containing nuclear-located Pax7 illustrated the following year [97]. Pax7 has become the most commonly used satellite cell marker, thanks to both its specificity and the excellent Kawakami monoclonal anti-Pax7 antibody (https://antibodyregistry.org - AB_528428) [102] available from the Developmental Studies Hybridoma Bank (DSHB) (https://dshb.biology.uiowa.edu/PAX7) that works in amphibian [103], bird [104] and mammal [9], including human [105, 106].
While no obvious abnormalities in myogenic precursor populations had been observed at embryonic stages in Pax7-/- mice [107], there was a striking phenotype in post-natal skeletal muscle of Pax7-null animals - namely, an absence of satellite cells and a lethal loss of myofibre diameter and muscle mass [9]. This established not only the importance of Pax7 in specification of the satellite cell pool, but also the role of Pax7-expressing satellite cells in muscle growth.
Crucially, the specificity of Pax7 to satellite cells in adult muscle initiated the use of the Pax7 locus to target satellite cells, for example with eGFP [39] for identification and isolation. Targeting the Pax7 locus with Cre-ERT e.g. [108] allowed satellite cell-specific conditional recombination, permitting knockout of genes and genetic ablation of satellite cells [1].
DISCOVERY OF THE MUSCLE REGULATORY FACTORS
The late 1980s marked discovery of the four myogenic regulatory factors (MRFs) MyoD, Myf5, myogenin and MRF4 (Myf6), an event that revolutionized understanding of developmental and regenerative skeletal myogenesis [10].
The critical observation by Taylor and Jones that a DNA methyltransferase inhibitor (5-azacytidine) could convert embryonic mouse fibroblasts to a variety of other lineages, including muscle [109, 110], was later attributed to demethylation of distinct regulatory loci [111, 112]. Stable heterokaryons made from fusion of diploid human amniocytes and differentiated mouse muscle cells showed that the human nucleus could be reprogrammed to express muscle genes, indicating ‘factor(s)’ transferring via the cytoplasm from the differentiated mouse muscle nucleus [21]. Such observations contributed to the hypothesis that expression of specific genes can reprogram cell fate.
Davis, Weintraub and Lassar generated a cDNA library of individual loci expressed in both C2C12 myoblasts and myoblasts converted from 5-azacytidine-treated fibroblasts, but absent in untreated fibroblasts [113]. A transcript was identified that, when transfected into 10T1/2 fibroblasts and other cell types, was sufficient to generate stable myogenic clones; the transcript was named Myogenic differentiation 1 (MyoD1, aka MyoD) [113].
Additional cDNA screening led to identification of myogenin due to high sequence homology to the myc domain of MyoD, reported separately by Wright, Sassoon and Lin [114] and Edmondson and Olson [115] in 1989. Myogenin facilitated myogenic conversion when transfected into non-muscle cells. Unlike MyoD though, endogenous myogenin was absent in proliferating myoblasts, but induced and peaked early in differentiation, shortly before fusion and expression of genes associated with muscle contraction. This led to speculation that myogenin directed the early skeletal muscle differentiation program.
Later in 1989, Braun et al. reported a third factor that they termed Myf5 [116], identified through screening transcripts expressed in foetal myoblasts that cross-hybridized with MyoD. Myf5 facilitated fibroblast-to-myoblast transdifferentiation with similar efficacy to MyoD [116].
MRF4 was the final member of the quartet, and entered the picture in September 1989 in a paper by Rhodes and Konieczny [117]. MRF4 was discovered through sequence homology to MyoD in a screen against an adult rat skeletal muscle library. MRF4 could convert 10T1/2 fibroblasts to the myogenic lineage and activate MyoD and myogenin in differentiating myotubes [117]. The human equivalent to MRF4 was then described, but called Myf-6 [118].
MUSCLE REGULATORY FACTOR EXPRESSION IN SATELLITE CELLS
During embryonic development, skeletal muscles of the head derive from unsegmented cranial mesoderm (comprising cranial, paraxial and precordal mesoderm). In contrast, the skeletal musculature of the body originates from transitory mesoderm-derived structures called somites, formed in a rostro-caudal progression in pairs flanking the neural tube [119, 120]. Somites differentiate into the dermomyotome and sclerotome, with mesodermal cells specified as muscle precursors in the nascent myotome. Migrating cells from the dermomyotome also provide myogenic precursors to form a portion of the tongue, some neck and jaw muscles, diaphragm, trunk and limb musculature [119, 120] and their associated satellite cells [121].
In situ hybridization showed that Myf5 and then MRF4 were first expressed in a population of cells in the murine dermomyotome which migrate to form the myotome. These cells subsequently express myogenin and differentiate as the first myogenic cells. MyoD expression in myogenic progenitors follows that of Myf5, MRF4 and myogenin [122–124].
In adult muscle, MRF4 was the most highly expressed [124, 125], but low levels of MyoD and myogenin were reported to correlate with fast and slow muscle fibre types respectively [126]. The crucial observation that MyoD and myogenin were redeployed during regenerative myogenesis was made in 1992 independently by Miranda Grounds and colleagues [127] (Fig. 3A) and Fuchtbauer & Westphal [128]. MyoD expression was noted in mononucleated cells in sections of regenerating muscle by 4–8 hours after acute crush injury, and then later in myotubes [127, 128]. These studies showed that the control of developmental and regenerative myogenesis employed similar regulatory pathways.
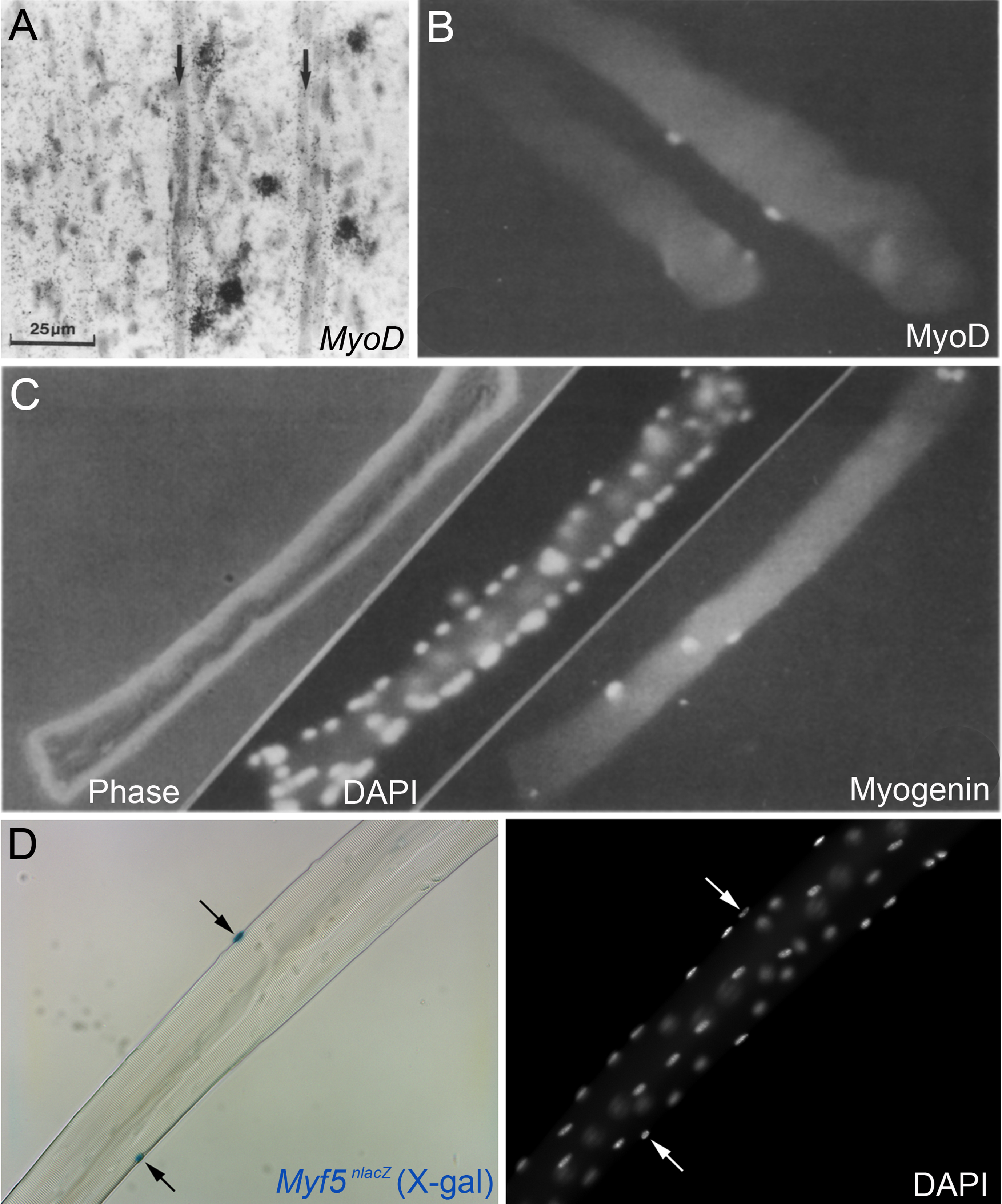
Myogenic regulatory factor expression in regenerating muscle and satellite cells. (A) In situ hybridization using a 35S-UTP labeled probe for MyoD in regenerating murine tibialis anterior muscle four days after crush injury, from Grounds et al., 1992 [127]. Hybridisation signal (silver grains) can be observed over some mononuclear cells but not over the newly formed myotubes (arrows). Scale bar represents 25μm. (B) MyoD immunolabeling of activated/proliferating satellite cells on an isolated myofibre from the flexor digitorum brevis of an adult rat after 2 days ex vivo, from Yablonka-Reuveni and Rivera, 1994 [52]. (C) Phase, DAPI nuclear counterstain and myogenin immunolabeling of a flexor digitorum brevis muscle fibre from an adult rat after 3 days ex vivo from Yablonka-Reuveni and Rivera, 1994 [52]. B and C illustrate that MyoD is followed by myogenin during myogenic progression in satellite cell progeny. (D) Isolated extensor digitorum longus myofibre from a Myf5nlacZ/+ mouse, with satellite cells localised by a blue precipitate from β-galactosidase activity from the nlacZ-targeted Myf5 allele following incubation in X-gal (left, arrows). DAPI counterstain (right) to visualize both satellite cell nuclei (arrows) and myonuclei, from Beauchamp et al., 2000 [15]. Images as originally published but panel labels have been amended/rearranged/added.
RT-PCR for MRF expression in plated adult rat satellite cells ex vivo revealed that MyoD was expressed first at around 12 hours and before proliferation, followed by Myf5 and MRF4 by 48 hours, and myogenin from 72 hours, concomitant with the first evidence of differentiation [129], with similar dynamics for MyoD and myogenin observed in Xenopus [130]. MRFs were not detected in quiescent satellite cells (although the Myf5 locus was later shown to be active [15]).
At the protein level, myogenic cells isolated from embryonic, foetal and newborn mice expressed the four MRFs with similar dynamics. MyoD appeared first followed by a transient wave of myogenin accompanying the onset of myotube formation, with MyoD and myogenin present in more cells than Myf5 or MRF4 [131]. Immunolabelling of satellite cells maintained in their niche on isolated myofibres ex vivo by Yablonka-Reuveni and Rivera elegantly showed the temporal progression of satellite cell myogenesis with MyoD and PCNA expressed within 2 days, then declining as myogenin and MyHC were expressed [52] (Fig. 3B and C). Interestingly, it was later shown that both Myf5 and MyoD levels were independently regulated through the cell cycle [132, 133].
These early studies provided the canonical markers and some of the antibodies still widely used to track the stages of regenerative myogenesis. Notable are monoclonal anti-MyoD clone 5.8A (AB_627978) [11], monoclonal anti-myogenin clone F5D (AB_2146602) [12, 134] and monoclonal anti-myosin heavy chain II clone MF20 (AB_2147781) [135], the latter two available from the DSHB (https://dshb.biology.uiowa.edu).
ASSESSING MUSCLE REGULATORY FACTOR FUNCTION USING NULL MOUSE MODELS
Gene inactivation studies followed to understand MRF function in vivo. The first model was the MyoD-null mouse, which was both viable and fertile, and developed overtly normal skeletal muscle but with an increased number of satellite cells [136]. However, the satellite cells in MyoD-null mice were initially thought incapable of carrying out regeneration following acute (crush) injury, which was further supported by the severity of the phenotype in a chronic muscle regeneration model (mdx/MyoD-null cross) [137]. Additional investigation suggested that MyoD-/- satellite cells underwent a prolonged period of expansion (but see [138]) and differentiated poorly in vitro [67, 140], however they were subsequently shown to be able to regenerate muscle in vivo after grafting, albeit at a slower pace [141].
Genetic inactivation of the other MRFs resulted in perinatal lethality, initially precluding analysis of MRF function in adult mice. Myf5-null mice died at birth due to rib cage abnormalities, but with no obvious skeletal muscle abnormalities [142]. Myoblasts isolated at E18.5 indicated that Myf-5-null satellite cells proliferated less and differentiated precociously in vitro [138]. When Myf5-null alleles were later made that allowed survival to adult, it was found that lack of Myf5 mildly affected satellite cell function [143, 144]. However, double Myf5/MyoD mutant mice had virtually no skeletal muscle [145], showing that one of either MyoD or Myf5 was required for skeletal muscle formation.
Parallel studies generated two myogenin-null mouse models, both of which had severe skeletal muscle defects and died perinatally [146, 147]. Myogenesis varied regionally, with muscle tissue containing disorganized myofibres and/or high quantities of mononucleated cells. Thus, myogenin was not necessary for myogenic specification, but played a pivotal role in muscle differentiation during development that could not be substituted by other MRFs. That being said, conditional inactivation of myogenin following embryonic muscle development showed that myogenin was dispensable for subsequent muscle growth, and so for satellite cell differentiation [148].
The situation with MRF4 was more complicated, with three different groups generating MRF4 knock-out mice; one of which was viable [149] while the other two died at birth [150, 151]. However, all three had varying reductions in Myf5 expression and resembled the Myf5-null models in terms of rib cage deformities. Since the MRF4 and Myf5 genes are close together on murine chromosome 10, it was speculated that the differences in phenotype were related to the degree to which targeting MRF4 affected the Myf5 locus in each model [152]. The viable MRF4-null model had apparently normal musculature in the adult [149] but any subtle effects on satellite cell homeostasis or muscle regeneration remain unknown [10].
These early knockout studies provided fundamental insight into myogenesis that also informed on the role of the MRFs in satellite cells. MRF function in adult has recently been comprehensively reviewed [10].
GENETIC MODIFICATION PROVIDES LINEAGE MARKERS FOR SATELLITE CELLS
An extremely helpful innovation by Shahragim Tajbakhsh in the Buckingham lab was to inactivate Myf5 by inserting a nlacZ reporter gene into the Myf5 locus to generate the Myf5 nlacZ allele [13]. This model was especially important in absence of useful Myf5 antibodies at the time, and allowed detailed examination of Myf5 expression and cell fate during development, as well as of the effects of loss of Myf5 function in null Myf5nlacZ/nlacZ mice [13]. Importantly, Myf5nlacZ/+ mice were viable, allowing high-resolution examination of Myf5 expression dynamics. Analysing Myf5 using the targeted Myf5 nlacZ allele, combined with immunolabelling for MyoD, during muscle regeneration in vivo permitted cellular resolution. This showed eventual co-expression of the two genes in most activated satellite cells and their myoblast progeny [153]. What came as somewhat of a surprise at the time though, was that the Myf5 nlacZ allele also revealed Myf5 expression in quiescent satellite cells in uninjured muscle [15] (Fig. 3D). This provided an easy high-throughput/high-resolution means to detect satellite cells in any mouse muscle [15].
Also in the Buckingham lab in Paris, a series of transgenes driving nlacZ had been generated in the mid 1990s by Robert Kelly to examine regulation of the sarcomeric MLC1/3F gene [14, 155]. Of these, the 3F-nlacZ-E transgene was robustly expressed in myonuclei [14] and so could also be used to identify satellite cells by their lack of transgene expression on isolated myofibres [15].
Importantly, it was realized that the 3F-nlacZ-E transgene could provide a sensitive lineage marker to determine if implanted cells contributed to host skeletal muscle [16, 156], while the Myf5 nlacZ allele could report contribution to the host satellite cell pool [16, 157].
GRAFTING STUDIES DEFINE SATELLITE CELL FUNCTION
Grafting has always been an important tool to examine muscle function, following its initial use by Volkmann in 1893 [5, 158]. Neerunjun and Dubowitz [159] and Snow [160, 161] dosed rodents with tritiated thymidine before transplanting muscles, showing that proliferating cells in regenerated muscles gave rise to labeled myonuclei. That these regenerative cells were myoblasts was confirmed by Lipton and Schultz, who labeled myoblast cultures with tritiated thymidine in vitro before grafting and detected label in myonuclei within host myofibres, but not in fibrogenic, endothelial or other non-muscle cell types [162]. Implantation of myoblasts under the skin also gave rise to myotubes [162].
Lineage tracing with tritiated thymidine however, had a number of drawbacks, including label dilution and laborious and indirect detection [122]. Having a traceable donor cell marker that was inherited only in donor-derived progeny was an obvious advantage, such as using the Y chromosome in male donor cells in female hosts e.g. [163]. Another strategy was grafting quail cells into chick embryos, which could then be identified in the host by virtue of their nuclear morphology [164]. This was the technique used by the Kieny group to show that satellite cells in the body, like their associated muscle fibres, originate from the somites [121].
An approach for mammalian systems employed by the Sloper group (including one Terence Partridge) used grafts between two strains of inbred mice, each homozygous for different allelic forms of the enzyme glucose-6-phosphate isomerase. This technique was used to show that donor cells could fuse with host cells or myofibres in adult muscle [165, 166].
Significantly, lineage tracing using isoenzymes also showed that after fusion, donor myogenic cells could introduce genes into the host muscle fibre. This led to the idea that functional versions of defective genes could also be delivered via cell fusion and would subsequently generate functional protein in genetically mosaic myofibres to treat inherited muscle disorders [167]. An appropriate model of a primary genetic muscular dystrophy was needed to test cell therapy in vivo [168, 169]. Such a model proved to be the mdx mouse that exhibited characteristics of muscular dystrophy [170]. When mutations in dystrophin were demonstrated to cause Duchenne muscular dystrophy [171], this led to identification of a nonsense mutation in exon 23 of the Dmd gene in mdx mice [172]. Partridge and colleagues showed that implantation of healthy myoblasts with a functional Dmd gene could confer dystrophin expression in host mdx muscle fibres [173]. There rapidly followed a series of cell therapy trials in boys with Duchene muscular dystrophy that highlighted highly challenging technical issues [169, 174]. Mdx mice had a comparatively mild dystrophic phenotype but a single dose of X-irradiation (16 Gy) to one leg inhibited regeneration to provide a more faithful dystrophic model and better recipient for grafting studies [175].
Grafted myoblasts could clearly form muscle and also fuse into existing muscle fibres, but a subpopulation of donor cells with stem cell-like characteristics could also be detected [176]. Labeling primary myoblasts with a retrovirus encoding nlacZ before grafting, Yao and Kurachi demonstrated that a small proportion of donor-derived cells persisted as myoblasts, which could then be recovered from host tissue and would differentiate ex vivo [177]. Crucially, recovered donor cells could also form muscle when re-implanted [177]. Use of H-2Kb-tsA58 myoblast clones showed that donor myoblasts could also participate in multiple rounds of host muscle regeneration [178], thus implanted myoblasts could reenter a quiescent, stem cell-like state, but remained capable of myogenesis. Importantly, labeled myogenic clones [179] and primary myoblasts from Myf5nlacZ/+ mice [157] were able to occupy the satellite cell niche following grafting. Later, transplantation of single muscle fibres from Myf5nlacZ/+ mice led to the demonstration that satellite cells were bona fide stem cells [16], with subsequent grafting studies confirming the classification [17, 180]. Interestingly satellite cells grafted when still near quiescence had better regenerative capacity [16, 180].
SATELLITE CELLS AS STEM CELLS
Much progress towards understanding how the satellite cell pool is maintained, as well as how such a small number of cells can replace large amounts of tissue so quickly, was also made during this era. The suggestion of asymmetric cell division had been made by Moss and Leblond in 1971, who proposed that half the satellite cell progeny fuse to the growing muscle fibres in 14–16 day old rats, while the other half divides again, before half of that generation again either differentiate or continue to divide, and so on [80]. This notion was reinforced by Quinn, Holtzer and Nameroff in the 1980s, who modelled myogenic cell commitment in vitro by quantifying clonal expansion of myogenic chick cells from 14 day embryos. In a series of papers, it was proposed that early myogenic precursor cells can divide either symmetrically into two stem-like progeny, or asymmetrically to produce one stem-like daughter cell that will continue to divide and one committed daughter cell that will divide a finite number of times (4) before differentiation, termed ‘quantal mitosis’. These observations suggested existence of a self-renewing stem cell in the skeletal muscle lineage [181–183]. Work on mouse and chicken muscle regeneration in vivo by Grounds and McGeachie supported the observation that some cells differentiated after only a few divisions while others underwent extended proliferation, but challenged the concept of quantal mitosis dictating a uniform, finite number of divisions of committed cells prior to fusion [184–186].
Myoblasts isolated from regenerating and control muscles could also be divided into two populations on the basis of cell size and colony size formed ex vivo [187]. Myoblasts with stem cell-like properties were also identified in human cultures [188]. Some myoblasts were also found to express myogenin inside 8 hours of muscle injury indicating rapid differentiation, whereas most did not even divide much before 24 hours [189]. In addition, two compartments were described in growing rat muscle with respect to their rate of cell division [190].
Whether or not satellite cells contain a distinct stem cell lineage is still an active debate [2, 19].
MODELING SATELLITE CELL MITOTIC QUIESCENCE
Satellite cell morphology was noted to change as muscle matures, with fewer ribosomes and less rough endoplasmic reticulum, indicative of reduced metabolic activity [80]. Edward Schultz built on earlier work [5] to show that satellite cells also became mitotically quiescent in adult mice, as they did not label with tritiated thymidine even after 9 days of continuous administration [191]. Once isolated, adult satellite cells were reported to not undergo cell division before 24 hours (unlike embryonic/foetal myoblasts), again testifying to their mitotically quiescent state in healthy adult muscle [29].
Keeping satellite cells fully quiescent during isolation is difficult [60], so means to induce quiescence in vitro provided a useful alternative. When human primary satellite cells or C2C12 cells are induced to differentiate in vitro, most form multinucleated myotubes, but a proportion of cells were observed to escape immediate differentiation to become ’reserve cells’: a slowly dividing or quiescent myogenic population. Upon stimulation, reserve cells would again divide rapidly and generate either differentiation-competent progeny, or further reserve cells [188, 192]. Significantly, reserve cells expressed markers characteristic of quiescent satellite cells such Myf5 and CD34 [15, 192]. Culture of satellite cells on isolated myofibres also demonstrated cells that returned to a quiescent state [193, 194]. Such models remain useful to explore myogenic quiescence and self-renewal.
SATELLITE CELL HETEROGENEITY
Studies on satellite cell proliferation and differentiation had indicated differences in cell behaviour, which prompted the question of whether satellite cells were a heterogeneous population. Certainly satellite cells from muscles with distinct embryonic origins were found to exhibit differences, such as expression of a cat jaw-specific superfast MyHC isoform in regenerated cat limb muscle only with prior grafting of jaw muscle-derived myogenic precursors [195, 196]. Similarly, proliferation of myogenic cells from the first branchial-arch derived masseter was poor compared to the tibialis anterior of the limb, and masseter (although not the non-somitic-origin digastric and sternocleidomastoid) also regenerated less effectively than tibialis anterior [197]. Heterogeneity in behaviour was also noted in satellite cells from within the same muscle, with the MyHC isoform repertoire upon differentiation reported to correlate with the phenotype of their associated myofibre in mouse [58] or muscle in chicken/quail [198].
SINGLE CELL TRANSCRIPTOMICS THE HARD WAY
Related to heterogeneity, in a pioneering study Cornelison and Wold examined gene expression in satellite cells at the single cell level [92]. Satellite cells were identified by first treating muscle fibres with Marcaine, a myotoxic drug that kills myofibres but spares satellite cells, and then collected from their niche following a live/dead stain. RNA was extracted from individual satellite cells at various time points post-isolation, and expression levels of C-Met, M-cadherin and the four MRFs assayed using multiplex single cell RT-PCR. C-Met was expressed in all quiescent satellite cells, while M-cadherin was only expressed in a fraction of quiescent satellite cells, with rare MRF expression detected. Following activation, satellite cells began to express either MyoD or Myf5, before eventually expressing both, followed by myogenin then MRF4 [92]. A follow up single cell analysis examined satellite cells from MyoD-null mice [140]. While single cell technology is all the rage these days e.g. [199, 200], the Cornelison and Wold [92] study was heroic.
NON-SATELLITE CELLS IN REGENERATIVE MYOGENESIS
Satellite cell function is supported and controlled by non-muscle cells [201, 202], and the 1990s saw the beginning of our understanding of how non-satellite cell types assist muscle regeneration. While invasion by neutrophils occurred first, macrophages were the main immune cells present by 2 days of muscle regeneration, with an initial population concerned with phagocytosis and clearing debris, before a later population emerged that supported muscle regeneration [203–205]. Indeed, co-culture of macrophages increased satellite cell-derived myoblasts proliferation and differentiation via secreted factors [206–208] and macrophages were found essential for muscle regeneration [209]. Further cell types supporting satellite cell function and muscle regeneration have subsequently emerged, such as fibro/adipogenic progenitors [210].
It is pertinent to note that the late 1990s also saw the claim of non-muscle cell or unorthodox sources for myogenesis. This had long been mooted, until the discovery of satellite cells seemingly defined the source of postnatal myoblasts [5]. Using the glucose-6-phosphate isomerase model, Grounds had reconstituted the bone marrow of irradiated mice, but could not detect a donor-derived contribution to muscle regeneration using Western blot to distinguish chimeric isoenzymes [211]. However, the single cell resolution afforded by the 3F-nlacZ-E transgene allowed Mavilio, Cossu and colleagues to spot individual skeletal muscle myonuclei after direct transplantation of donor bone marrow from 3F-nlacZ-E mice into regenerating host wild-type mouse muscle [156]. Donor cell recruitment into regenerating host muscles following bone marrow transplantation into lethally-irradiated or non-irradiated host animals was also reported [156, 163].
A population of bone marrow cells could be purified due to their relative ability to exclude Hoechst dye, which was thought to include the most primitive haematopoietic stem cells; a population that was called side population or SP cells [212]. Intravenous injection of either wild-type haematopoietic stem cells or bone marrow-derived SP cells into irradiated mdx mice gave reconstitution of the haematopoietic lineage, as well as donor-derived nuclei in host muscle as detected by donor-derived dystrophin expression [213]. Pericytes from the embryonic aorta and other vessels were also able to contribute to skeletal muscle upon grafting using the 3F-nlacZ-E mouse [214].
Other potential unorthodox sources of muscle followed over the years [215], but modifying the Pax7 locus to genetically ablate satellite cells showed that such unorthodox cells were unable to replace satellite cell function in muscle regeneration [1]. Nevertheless, alternative sources of muscle continue to be explored for therapeutic value e.g. [216]. Techniques to direct induced pluripotent stem cells [217] towards the skeletal myogenic and satellite cell lineages [218] certainly fall under the ‘non-muscle or unorthodox sources for muscle’ moniker.
SATELLITE CELL MEETINGS
Arguably, another foundation in satellite cell research initiated last century was establishment of the FASEB Satellite Cell Meetings! In 1969, the Muscular Dystrophy Associations of America Inc sponsored the first international conference on muscle regeneration, chaired by Alexander Mauro at the Institute for Muscle Disease in New York. The proceedings of the meeting were published the following year in a book titled ‘Regeneration of Skeletal Muscle and Myogenesis’, edited by Alexander Mauro, Saiyid Shafiq and Ade Mihorat [219]. This was followed in 1978 by a second conference on muscle regeneration at the Rockefeller University in New York, with the papers again collated in book form edited by Alexander Mauro in 1979, and simply called ‘Muscle Regeneration’ [220]. Mauro’s final contribution to the satellite story was his 1988 paper reporting no tight electrical coupling between satellite cells and their associated muscle fibre [221], before his death in October 1989 (his obituary in the New York Times mentions him as co-inventor of one of the first cardiac pacemakers, but no mention of satellite cells!). Satellite cells also featured in the Muscular Dystrophy Association International Conference on Myoblast Transfer Therapy in 1989, again in New York [222].
Satellite cell research did not really feature prominently in Myogenesis meetings of the 1990s, which were dominated by research focused around muscle development. This prompted a push for a conference again dedicated to satellite cells and postnatal myogenesis, and a steering committee of Judy Anderson, Ron Allen, Steve Hauschka, Orna Halevy, Miranda Grounds, Gillian Butler-Browne, Anna Starzinski-Powitz and Zipora Yablonka-Reuveni formed during the 1997 ‘Molecular Biology of Muscle Development’ Keystone meeting. Judy Anderson and Ron Allen then largely organized and co-chaired that first meeting called ‘Post-natal Myogenesis: Satellite Cells in Action!’ in August 1998 in Boston, USA, which also instigated the ‘The Alexander Mauro Lecture’, first given by Richard Bischoff (the program and announcements are included here as supplementary information). The subsequent nine meetings, starting in 2001, have been organized under the auspices of FASEB and remain a central platform for the skeletal muscle and satellite cell community.
SUMMARY
By the turn of the century then, the foundations in knowledge, tools and technology had been laid [5] (Fig. 4) to support satellite cell research in the 21st century. While the depth and nuance of our understanding of satellite cell function and muscle regeneration has greatly advanced in recent years [2, 19], these studies remain central pillars of regenerative myogenesis.
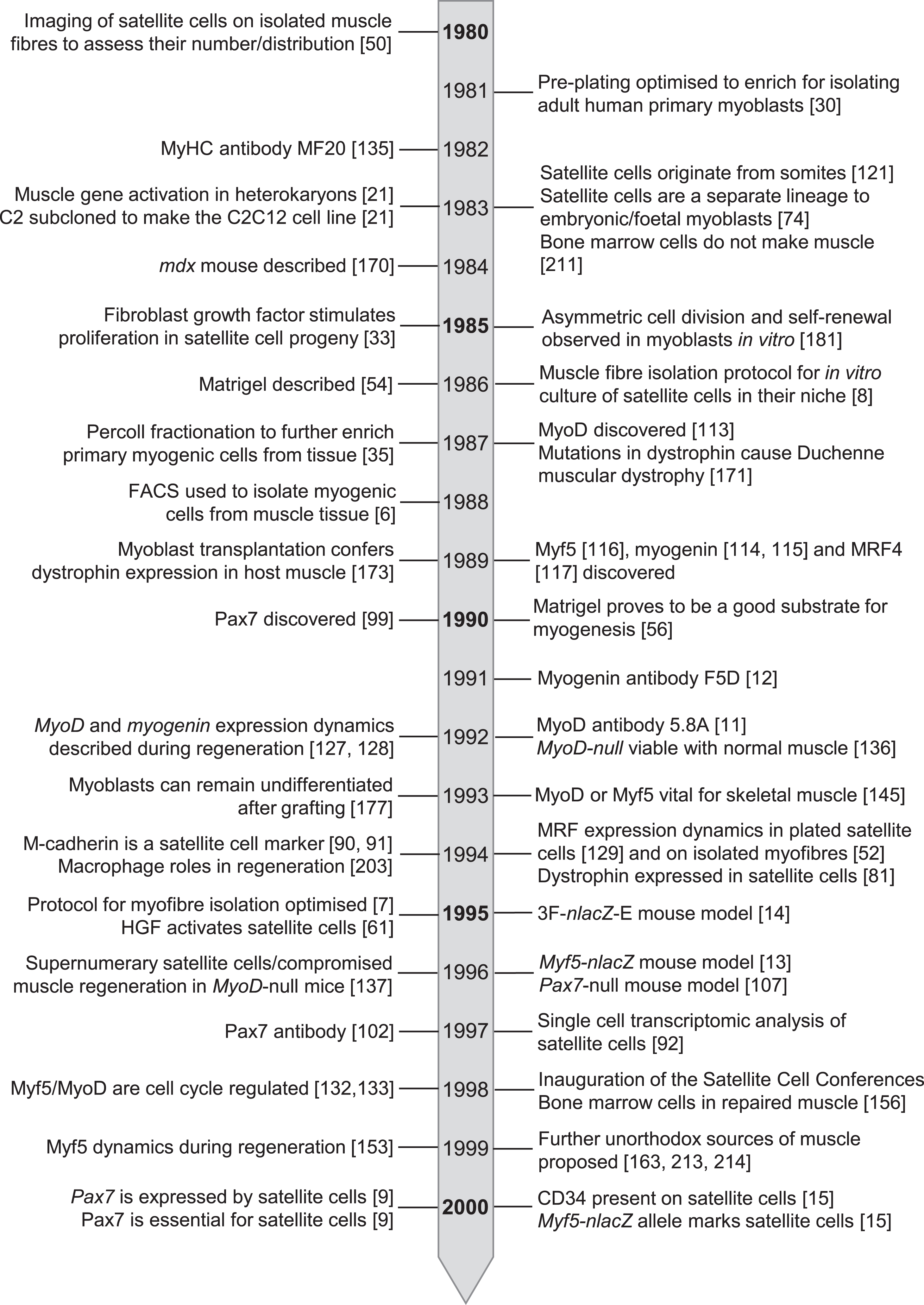
Timeline of seminal events in satellite cell research from 1980 – 2000.
Footnotes
ACKNOWLEDGMENTS
This review is dedicated to the boundlessly inquisitive and enthusiastic Terence A. Partridge on the occasion of his 80th Birthday. Thanks to Zipora Yablonka-Reuveni and Judy Anderson for valuable feedback and to Dawn Cornelison for finding the autographed 1998 program. Elise Engquist is supported by Wellcome Trust 222352/Z/21/Z from 108874/B/15/Z. The Zammit lab is supported by grants from the Medical Research Council to P.S.Z. (MR/P023215/1 and MR/S002472/1), from Muscular Dystrophy UK (RA3/3052), Association Française contre les Myopathies (AFM 17865) and the FSH Society (FSHS-82013-06 and FSHS-82017-05). Images are reproduced from published papers as indicated and with permission: Figure 1A - License Number 5057590765121, Fig. 1B - License Number 5057610080932, Fig. 1C - License Number 5057611172253 (at a cost of £75.65 – thanks Springer-Nature), Fig. 2A - License Number 5045410222306, Fig. 2B - License Number 5045410473405, Fig. 2C and 2D - License Number 5045410713875, Fig. 2E and F - Order license ID 1113708-1, ![]() A - License Number 5058860350012 (with another £75.65 to Springer-Nature!) and Fig. 3B and C - License Number 5052430512923.
A - License Number 5058860350012 (with another £75.65 to Springer-Nature!) and Fig. 3B and C - License Number 5052430512923.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.