
Abstract
Background:
Spinal Muscular Atrophy (SMA) is an inherited neurodegenerative disease caused by the loss or mutation of the survival motor neuron 1 (SMN1) gene. Though classically regarded as a motor neuron disorder, reports are increasingly describing the involvement of non-neuronal organs in SMA. The Smn2B/− mouse is a model of SMA that displays a peripheral phenotype including metabolic defects.
Objective:
Here, we characterized several neuronal and non-neuronal defects in the Smn2B/− mouse throughout development to better understand the progression of the disease and the relationship between tissue defects.
Methods:
We collected tissues from mutant Smn2B/− mice and Smn2B/+ littermate controls at several timepoints and evaluated spinal cord motor neuron loss, neuromuscular junction pathology, muscle fiber size, liver steatosis, and pancreatic islet cell composition. Blood glucose and plasma neurofilament light chain (NfL) were also measured.
Results:
Smn2B/− mice displayed several peripheral defects prior to motor neuron loss and showed early elevations in neurofilament light chain (NfL) protein.
Conclusions:
This work provides an important framework for guiding future research with this mouse model and demonstrates that the liver may be an early target in the development of SMA.
Keywords
Introduction
Spinal Muscular Atrophy (SMA) is a deadly childhood neurodegenerative disorder characterized by the wasting of skeletal muscle due to lower motor neuron death. The worldwide prevalence of SMA is estimated to be 1–2 in 100,000 individuals, with an incidence of about 1 in 12,000 live births.1,2 This inherited disease is caused by a homozygous deletion or mutation of the survival of motor neuron 1 (SMN1) gene. SMN1 produces the survival motor neuron (SMN) protein, which is required for motor neuron development and maintenance.3,4 The closely similar SMN2 gene produces only a small amount of full-length SMN protein and is unable to fully compensate for the loss of SMN1. 5 SMN2 gene copy number varies between individuals, and can thus influence the severity of disease in patients with SMA. 6 Other factors, such as modifier genes, may also play a role in influencing the severity of disease.
While patients with severe SMA previously had a life expectancy of less than 2 years, they now have access to several life-prolonging treatments. Three drugs: nusinersen, risdiplam and onasemnogene abeparvovec, are currently authorized for use by the United States Food and Drug Administration (FDA) and European Medicine Agency (EMA), while several other therapies are in pre-clinical and clinical development. Interestingly, therapeutic strategies are more effective when delivered systemically instead of directly to the central nervous system, likely due to the contributions of peripheral organs to SMA disease.7–9
SMA has been historically studied as a motor neuron disorder. However, SMN is a ubiquitously expressed protein and a growing body of evidence describes the effects of SMN depletion in non-neuronal tissues. Patients with SMA report higher instances of cardiovascular, gastrointestinal, metabolic, reproductive, and skeletal defects than healthy peers, which occur prior to the first signs of neuromuscular disease. 10 Further, mouse models of SMA have been used to characterize the involvement of non-neuronal organs in SMA. The Smn2B/− mouse has a genetically modified mutant Smn allele that produces only a small amount of SMN protein, similar to the human SMN2 gene.11,12 This model represents a severe SMA phenotype, displaying motor neuron degeneration, muscle weakness, and neuromuscular junction pathology. These mice display motor function deficits starting at postnatal day 9 (P9), and generally live for a median of 25 days.11,13 Interestingly, several non-neuronal phenotypes have also been described in Smn2B/− mice including immune organ defects, gastrointestinal dysfunction, liver steatosis, and pancreatic pathology.14,15 While it is becoming clear that the depletion of SMN affects several systems within the body, the relationship between the affected systems and their impact on the overall development of disease in SMA is not well understood.
To better outline the relationship between affected tissues and their contributions to disease development, we characterized several neuronal and non-neuronal phenotypes in Smn2B/− mice throughout development. We show that several non-neuronal symptoms arise before motor neuron loss, and that liver pathology occurs early in disease progression.
Materials and methods
Animals
Smn2B/− and Smn2B/+ mice were generated by crossing Smn+/− and Smn2B/2B mice (both on the C57BL/6J genetic background). All mice were housed at the University of Ottawa Animal Facility and experimental protocols were approved by the Animal Care Committee of the University of Ottawa. Animals were cared for in accordance with the guidelines of the Canadian Council on Animal Care, and the Animals for Research Act. One litter of mice was monitored daily for 60 days for survival, while additional litters were weighed and sacrificed for blood and tissue collection at P5, P7, P9, P11, P13, P15, P17, and P19 (Figure 1(A)). Four to five mice were used for each genotype at each time point. For plasma NfL concentration assays, blood was taken at P4, P7, P11, P15, and P19. Five to ten mice were used at each time point.
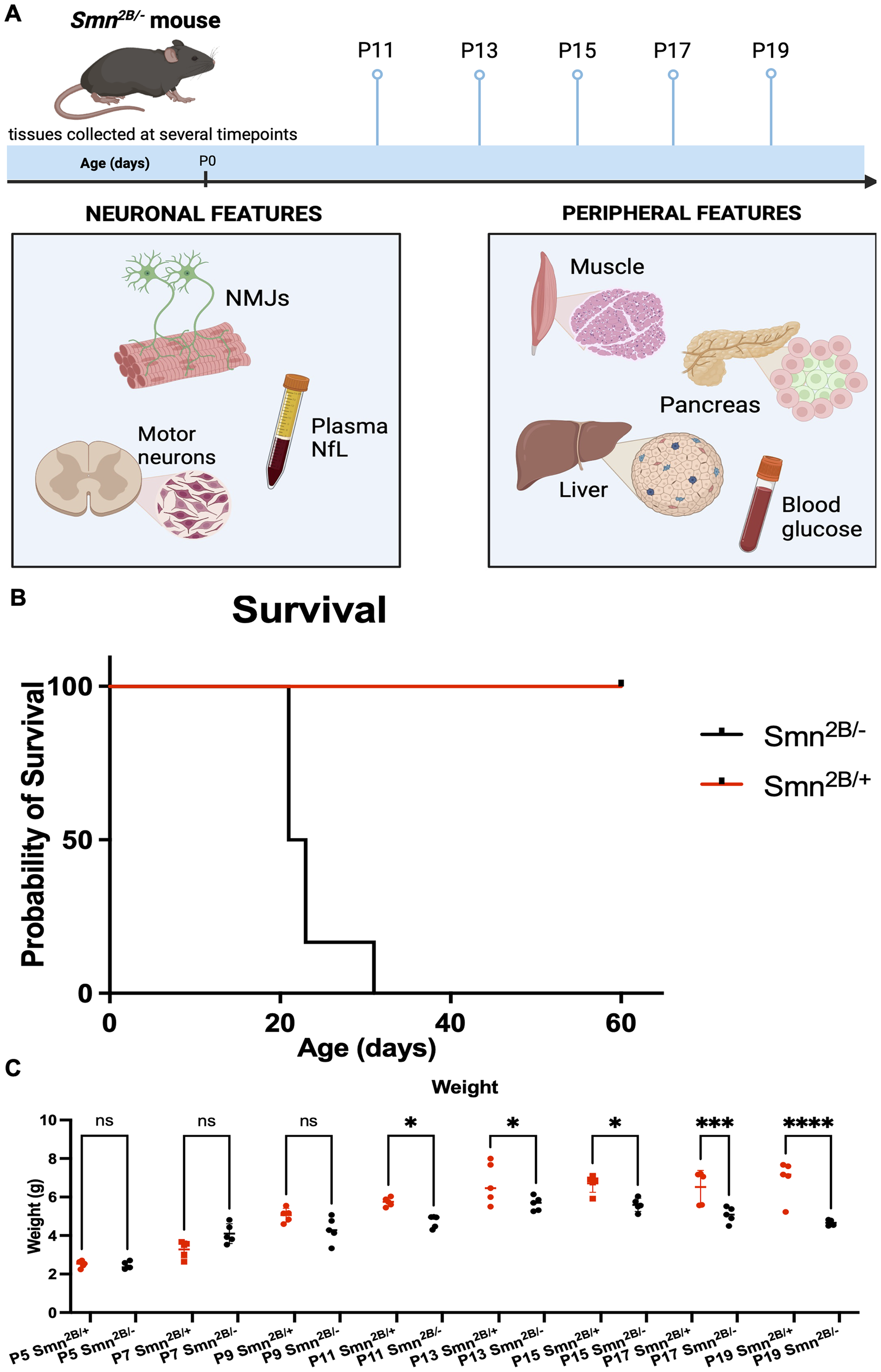
Weight and survival of Smn2B/− mice throughout development. (A) Schematic representation of experimental design for temporal evaluation of SMA-like characteristics in Smn2B/− mice. (B) Kaplan-Meier survival curve comparing Smn2B/− and Smn2B/+ mice up to 60 days. (C) Comparison of weights between Smn2B/− and Smn2B/+ mice from P5 to P19. Created with BioRender.com
Tissue processing and staining
Spinal cords were collected and choline acetyltransferase (ChAT) immunostaining was performed as previously described to visualize motor neurons.13,16 For each animal, 10 sections were stained and 20 ventral horn regions were quantified by the counting of ChAT positive motor neurons. Transversus abdominis muscle was collected, stained with TRITC-conjugated bungarotoxin, and immunostained for neurofilament and synaptic vesicle protein to visualize neuromuscular junctions as before. 13 Endplates were marked as either positive or negative for displaying neurofilament accumulation and/or denervation. Liver and tibialis anterior muscle were processed and stained with hematoxylin and eosin (H&E) as previously described. 13 The average cross-sectional area of 100 muscle fibers was measured using NIH ImageJ to quantify muscle fiber size. Pancreas was collected and immunostained for insulin and glucagon to visualize alpha and beta cells of the pancreatic islets as before. 13 Cells from at least 5 islets per animal were counted as either insulin or glucagon positive to establish an average ratio of alpha to beta cells. Antibodies used are listed in Table 1.
List of antibodies used for immunohistochemistry.
Blood collection and plasma analysis
Blood samples were collected upon euthanasia. Plasma was extracted and NfL protein concentration was determined using the Simoa NF-Light® assay (Quanterix, Billerica, MA) on a Simoa HD-1 analyzer as previously described. 17 Blood glucose was measured as before using a Freestyle Precision Neo meter (Abbott, Lake County, IL). 13
Statistical analysis
Data are presented as the mean ± standard error of the mean. Unpaired t-tests were used to compare the means of each genotype at each time point. Samples sizes were either 4 or 5 mice, except for the blood NfL tests where 5 to 10 mice were used for each time point. Significance was indicated by * for P ≤ 0.05, ** for P ≤ 0.01, and *** for P ≤ 0.001.
Results
Smn2B/− mice survive 22 days and show decreased weight gain starting at P11
To characterize the progression of disease in the Smn2B/− mouse, we first performed a survival analysis and measured weight over time in comparison to Smn2B/+ littermates. Smn2B/− mice showed a median survival of 22 days while all Smn2B/+ mice survived the 60-day monitoring period (Figure 1(B)). The first evidence of the diseased phenotype arises at P11, when Smn2B/− mice are maller in weight than Smn2B/+ controls. Following this, Smn2B/− mice begin to lose weight and Smn2B/+ continue to gain weight (Figure 1(C)). As this timepoint is the first evidence of the SMA-like phenotype in the Smn2B/− model, subsequent characteristics were evaluated beginning at P11.
Motor neuron degeneration begins at P19 in Smn2B/− mice and Nfl is elevated at P11
We next measured the degree of motor neuron degeneration, a hallmark feature of SMA, in Smn2B/− mice throughout development. Motor neuron number was determined by ChAT immunostaining of motor neuron cell bodies within the anterior horn of the lumbar spinal cord. Motor neuron cell body number was lower in Smn2B/− than Smn2B/+ mice beginning at P19 (Figure 2(A) and (B)). We also measured plasma NfL, a neuronal protein that shows potential as a biomarker of axonal damage and disease progression in SMA.18,19 Plasma NfL was first elevated in Smn2B/− mice at P11, several days earlier than the first appearance of motor neuron degeneration (Figure 2(C)).
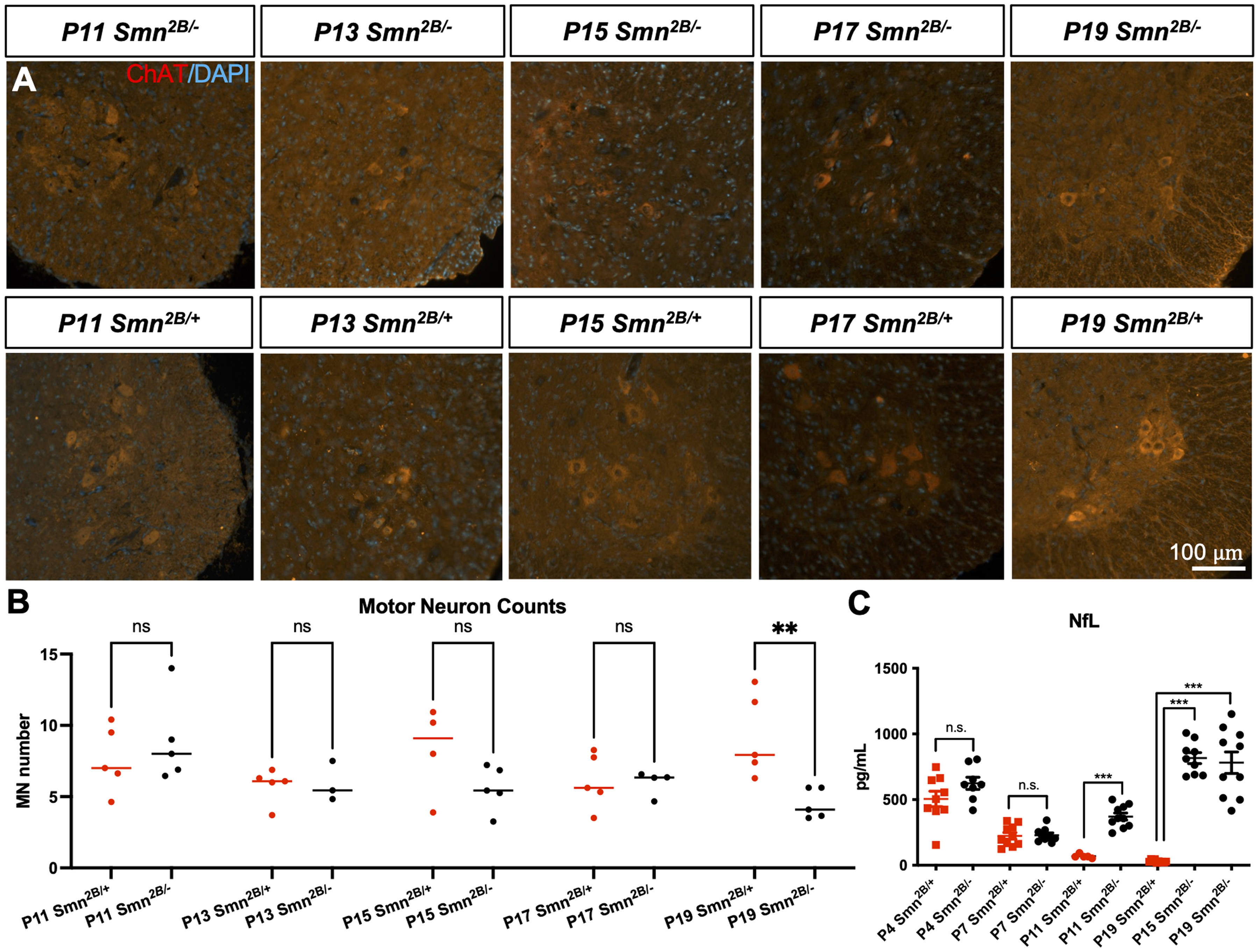
Temporal analysis of motor neuron degeneration in Smn2B/− mice. (A) Representative immunofluorescent images of sections of lumbar spinal cord anterior horns from Smn2B/− mice stained for ChAT (red) and DAPI (blue). (B) Quantification of motor neuron cell body numbers. (C) Plasma NfL levels. (n = 4-10, mean ± SEM, individual t-tests at each time point, p ≤ 0.01 for **, p ≤ 0.001 for ***). Scale bar = 100 μm.
Presynaptic neurofilament protein accumulation is first apparent at P13 in Smn2B/− mice, and denervation is apparent at P17
We next assessed neuromuscular junction (NMJ) pathology to further investigate how motor neurons are affected throughout development in the Smn2B/− model. Smn2B/− mice show defects at the site of the NMJ, including altered endplate morphology, accumulation of neurofilament protein at the nerve terminal, and denervation of endplates. 20 Upon investigation of the NMJs of the transversus abdominis muscle through neurofilament and synaptic vesicle protein immunostaining, presynaptic neurofilament protein accumulation was first apparent at P13, while denervation of endplates was first apparent at P17 (Figure 3). NMJs are therefore affected prior to the loss of motor neuron cell bodies in the spinal cord.
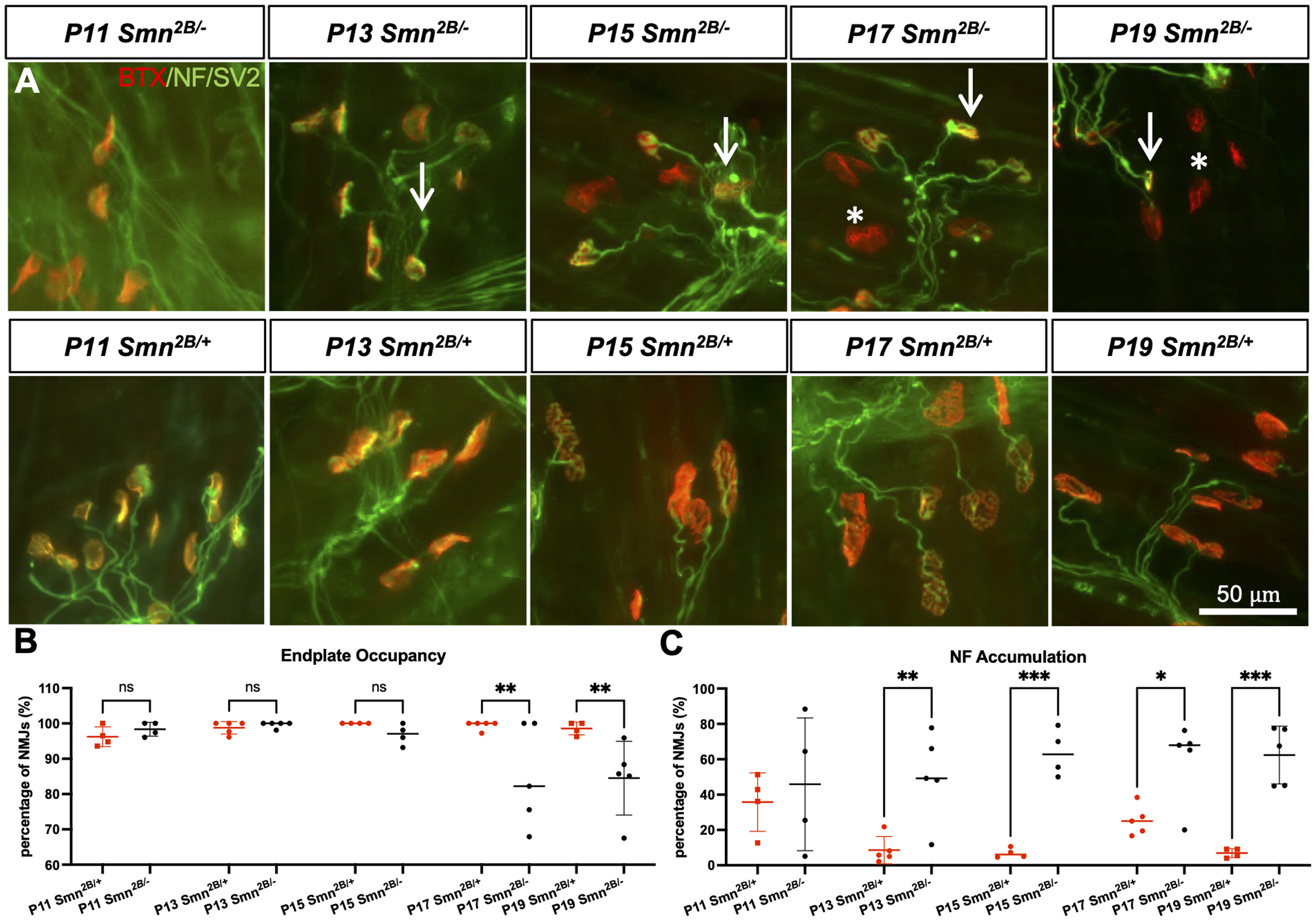
Temporal analysis of neuromuscular junction pathology in Smn2B/− mice. (A) Representative immunofluorescent images of transversus abdominis (TVA) muscle from Smn2B/− mice stained with bungarotoxin (red), and for neurofilament (NF) (green) and synaptic vesicle protein 2 (green). Quantification of endplate occupancy (B) and neurofilament accumulation (C) in stained NMJs (asterisks depict unoccupied endplates; arrows indicate NF accumulations; n = 4–5, mean ± SEM, individual t-tests at each time point, p ≤ 0.05 for *, p ≤ 0.01 for **, p ≤ 0.001 for ***). Scale bar = 50 μm.
Cross-sectional muscle fiber area is reduced in tibialis anterior muscle from Smn2B/− mice starting at P17
To determine when muscle atrophy is first occurring in Smn2B/− mice, we measured the cross-sectional area of H&E-stained muscle fibers from the tibialis anterior muscle. Muscle atrophy is another hallmark characteristic of SMA, likely caused by the degeneration of motor neurons as well as intrinsic mechanisms affecting muscle development.21–23 Muscle fiber size was first reduced in Smn2B/− mice compared to Smn2B/+ mice starting at P17 (Figure 4). Muscle atrophy thus coincides with denervation at P17, following neurofilament accumulation at the NMJ at P13 and preceding motor neuron degeneration in the spinal cord at P19.
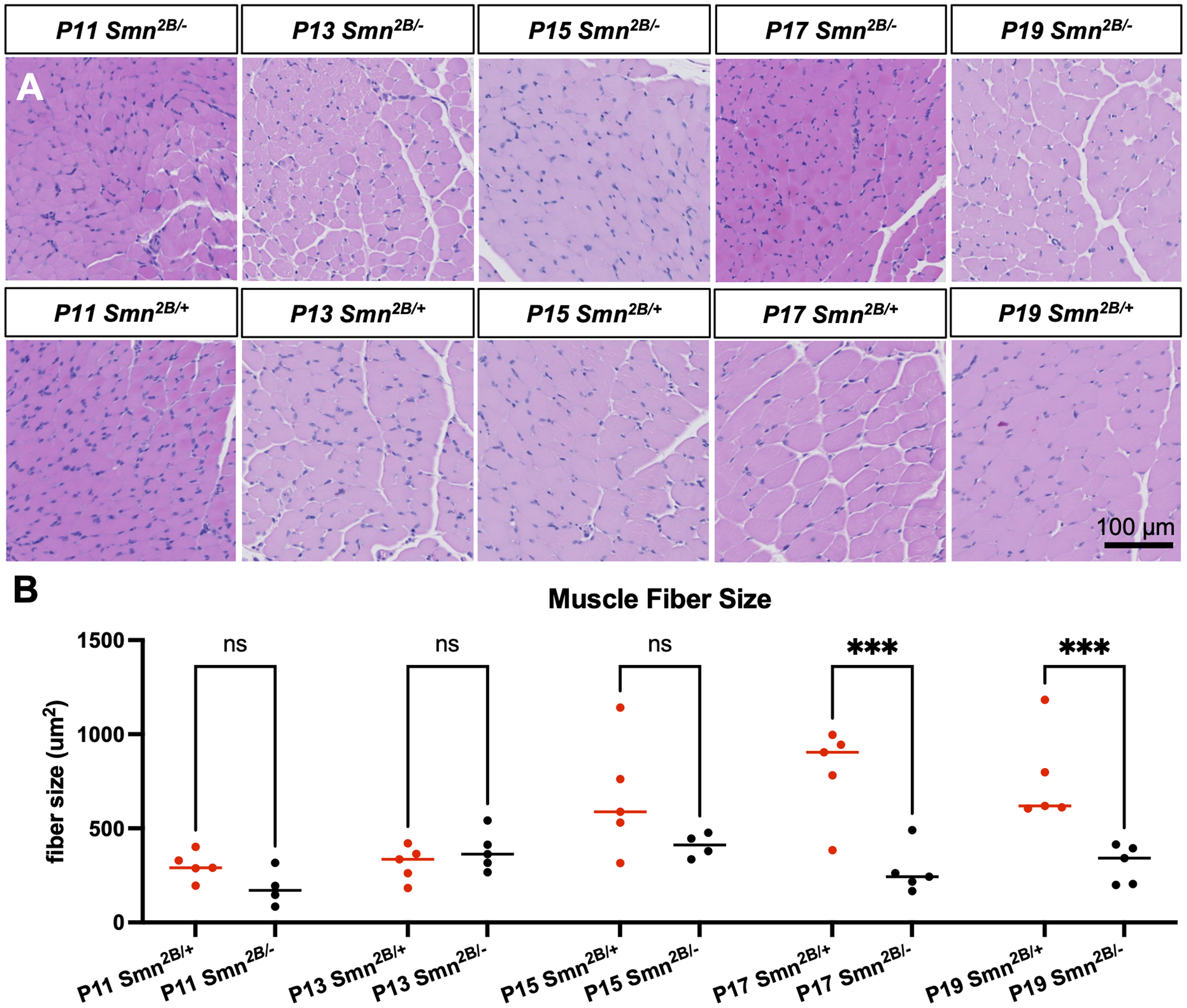
Temporal analysis of muscle fiber size in Smn2B/− mice. (A) Representative images of H&E stained tibialis anterior sections from Smn2B/− mice. (B) Quantification of muscle fiber size (n = 4–5, mean ± SEM, individual t-tests at each time point, p ≤ 0.001 for ***). Scale bar = 100 μm.
Liver steatosis is first apparent at P13 in Smn2B/− mice
In addition to neuromuscular pathology, Smn2B/− mice display an extensive number of metabolic abnormalities including liver steatosis and dysfunction.14,24 Steatosis was qualitatively observed using H&E-stained liver cross sections to determine the onset of the development of liver pathology in Smn2B/− mice. Fat was first observed within hepatocytes of Smn2B/− mice starting at P13 (Figure 5). The liver is therefore affected early in the Smn2B/− model, prior to neuromuscular pathology.
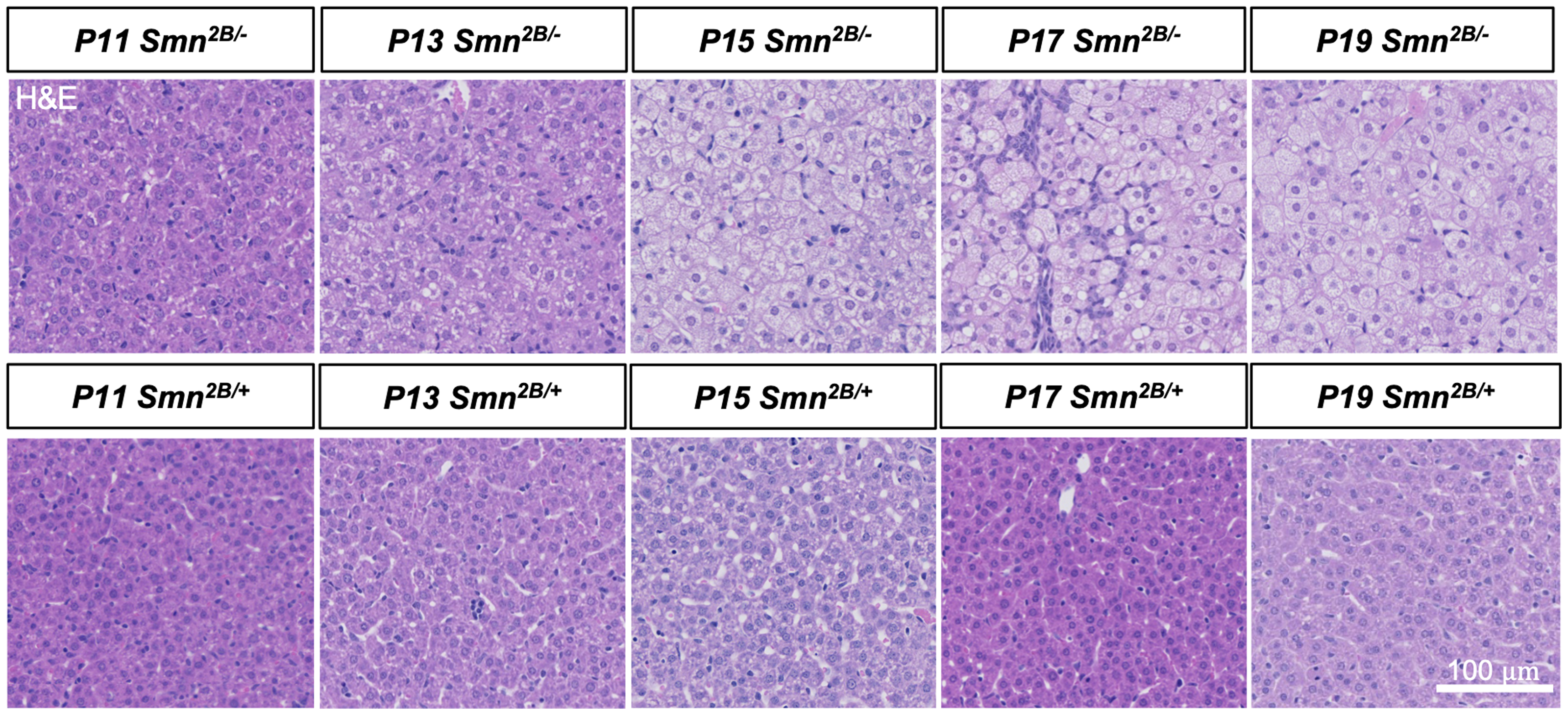
Temporal analysis of liver steatosis in Smn2B/− mice. Representative images of H&E stained liver sections from Smn2B/− mice. Scale bar = 100 μm.
Ratio of alpha to beta cells in pancreatic islets is disrupted beginning at P19
Smn2B/− mice display glucose metabolism defects including hyperglucagonemia, glucose resistance, and an increased ratio of glucagon-producing alpha cells to insulin-producing beta cells in the pancreas. 15 To determine the onset of pancreatic pathology during development, pancreata were immunostained for insulin and glucagon and the ratio of alpha to beta cells within the pancreatic islets was measured. The ratio of alpha to beta cells was first disrupted in Smn2B/− mice at P19 (Figure 6). Blood glucose was also measured as another representation of glucose metabolism defects in the Smn2B/− model. Blood glucose was decreased in Smn2B/− mice compared to Smn2B/+ mice starting at P15 (Figure 7). Glucose metabolism defects thus likely precede pancreatic pathology, as does liver steatosis.
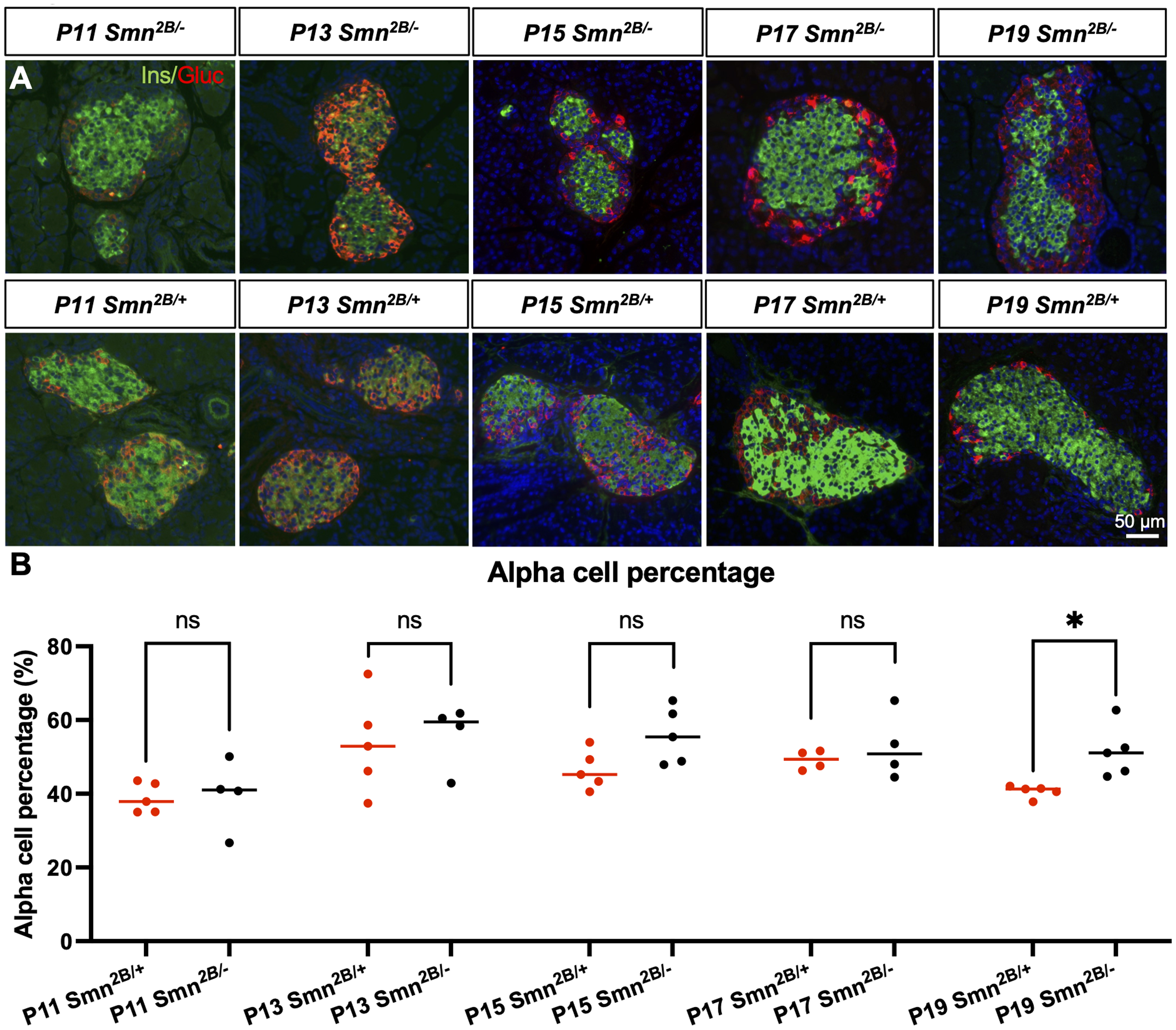
Temporal analysis of pancreatic pathology in Smn2B/− mice. (A) Representative immunofluorescent images of sections of pancreatic islets from Smn2B/− mice stained for glucagon (red) and insulin (green). (B) Fraction of glucagon-positive alpha cells related to total number of pancreatic islet cells (n = 4–5, mean ± SEM, individual t-tests at each time point, p ≤ 0.05 for *). Scale bar = 50 μm.
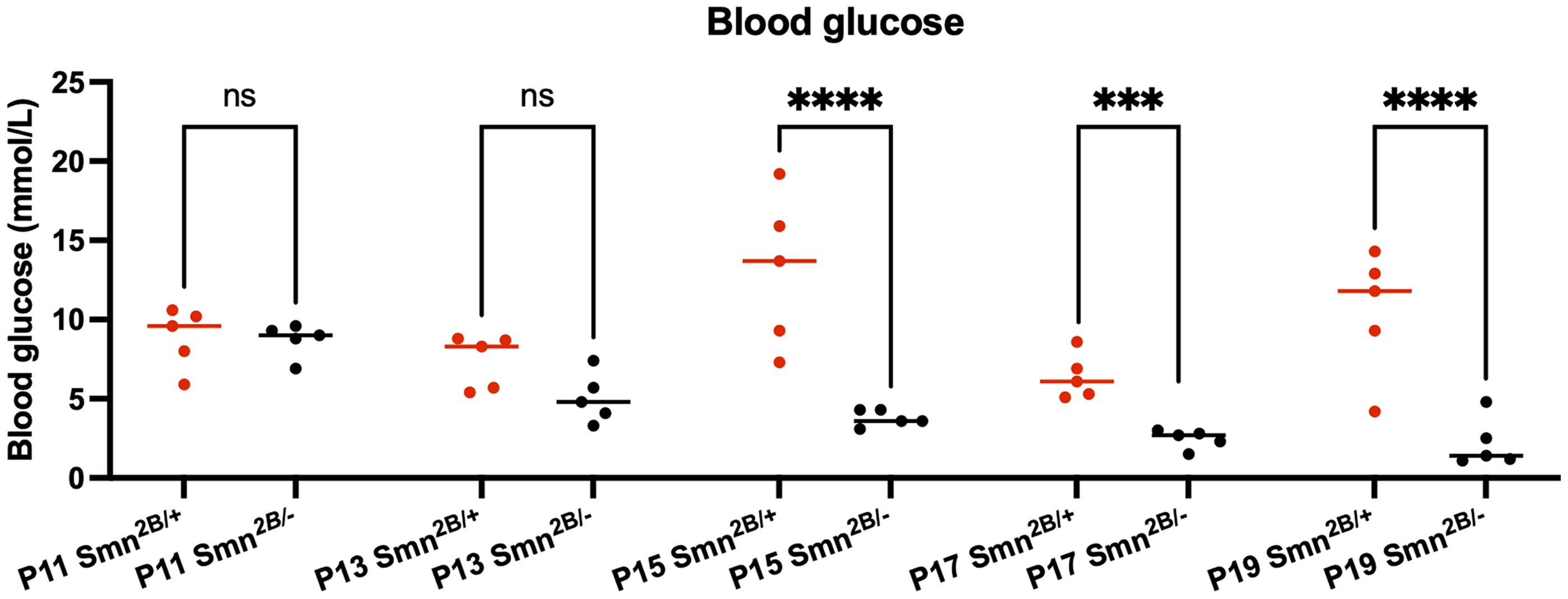
Temporal analysis of blood glucose in Smn2B/− mice. Quantification of blood glucose levels in Smn2B/− mice over time. (n = 5, mean ± SEM, individual t-tests at each time point, p ≤ 0.001 for ***, p ≤ 0.0001 for ****).
Discussion
We have presented an in-depth temporal characterization of the Smn2B/− mouse model of SMA. While many of these characteristics have been previously described in Smn2B/− mice,11,14,15,24 this is the first study to identify the initial occurrence of each phenotype during development. Mice show slowed growth beginning at P11, coinciding with elevated NfL protein in plasma. They display liver steatosis and neurofilament accumulation at the endplates of neuromuscular junctions starting at P13. Muscle fiber size and blood glucose are decreased from P15 onwards. NMJ endplates are denervated starting at P17. Loss of motor neuron cell bodies in the spinal cord and a disruption of the ratio of alpha to beta cells in the pancreas are seen at P19. These mice survive to a median of 22 days (summarized in Figure 8).
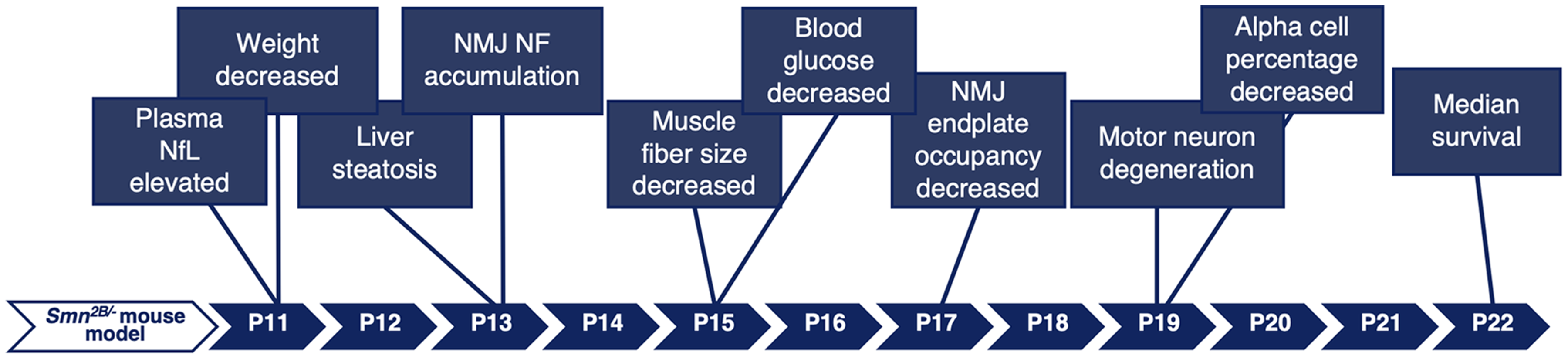
Graphical summary of the age of onset of SMA-like characteristics in Smn2B/− mice.
Smn2B/− mice display a non-alcoholic fatty liver disease (NAFLD) phenotype, consisting of steatosis, liver damage, liver dysfunction, and dyslipidemia at P19.14,24 Here, we show the emergence of liver steatosis early in the development of the disease, at P13, several days prior to motor neuron degeneration. These results support findings that liver pathology occurs prior to neuromuscular symptoms, though a previous finding identified fat accumulation even earlier, at P9. 14 The mechanism of the liver phenotype is unclear, though its early emergence would suggest that it is the result of SMN-dependent molecular pathways within the liver. The pancreas was previously theorized to be implicated, due to the unusually high number of glucagon-producing alpha cells and high levels of blood glucagon.15,24 However, we showed that the alpha/beta cell ratio was altered late in Smn2B/− mice, several days after the emergence of the liver phenotype. In fact, the pancreatic cell ratio could be a result of metabolic stress, as beta cells are more prone to death than alpha cells when exposed to hyperlipidemia. 25 This is supported by the impacts of a liver-specific depletion of SMN, which revealed pancreatic defects similar to those in Smn2B/− mice, emphasizing the liver's impact on the pancreas in this mouse model (de Almeida et al., unpublished data). Further, glucagon resistance and hyperglucagonemia are known to be associated with NAFLD. 26 High glucagon could thus be a result of liver steatosis rather than a contributing factor. More insight into the liver-intrinsic nature of these defects was provided by a study describing a liver-directed restoration of SMN in Smn2B/− mice. An adeno-associated virus-mediated restoration of SMN restricted to the liver provided a rescue of the fatty liver phenotype and pancreatic defects (Sutton et al., unpublished data). The liver-directed treatment also partially protected survival, motor function and NfL protein levels, emphasizing the influence of the liver on neuronal development through the regulation of neurotrophic factors like IGF1. 27
We also showed that liver steatosis occurs prior to NMJ denervation and muscle atrophy in Smn2B/− mice. This agrees with previous studies, which found no steatosis in livers from mutant SOD1G93A mice, a model of ALS with similar denervation and wasting of skeletal muscle. 14 Steatosis is therefore likely not the result of denervation or reduced muscle use. In fact, hyperlipidemia associated with the fatty liver phenotype could affect muscle function and exacerbate muscle atrophy through lipotoxicity. This is supported by a previous study that identified lipid droplets on muscles from Smn2B/− mice. 28
Muscle atrophy begins before motor neuron loss in Smn2B/− mice, likely due to the involvement of muscle intrinsic pathways and NMJ defects. Intrinsic defects in muscle development have been observed previously in SMN depleted myoblast cultures, myoblasts from patients with SMA, and in muscle-specific SMN knockout mouse models.21–23 Muscle atrophy was also preceded by neurofilament accumulation at the NMJ, indicating that neurofilament accumulation could be an early contributor to muscle atrophy, preceding denervation and the loss of motor neuron cell bodies. However, the consequences of neurofilament accumulation at the nerve terminal are unclear. Accumulations are present in denervation resistant muscles from SMA mice, suggesting that it may not be the cause of denervation. 29 Neurofilament accumulations could nevertheless affect the function of the NMJ or be representative of other issues with axonal transport within the neuron.
Neurofilament protein is also being explored as a potential biomarker for SMA. Neurofilament light chain (NfL) protein is elevated in plasma and CSF of pediatric SMA patients and is believed to act as a marker of neuronal degeneration.18,19 Here, we provide the first systematic characterization of plasma NfL throughout development in Smn2B/− mice. We observed progressive elevations of NfL that increased with the development of the neuromuscular phenotype. However, the first NfL elevations were observed prior to any of the pathological findings, suggesting that NfL could also be useful as a prognostic biomarker. Interestingly, NfL was elevated several days prior to denervation and motor neuron degeneration. These elevations could be representative of axonal transport dysregulation and NMJ neurofilament accumulation rather than motor neuron degeneration. NfL levels could also be elevated as a reflection of other neuronal populations. Astrocytes, microglia, interneurons, and sensory neurons have all been shown to be affected in animal and cells models of SMA.30–33 Alternatively, the NfL assay may be a more sensitive method for measuring motor neuronal degeneration than immunohistochemistry.
The Smn2B/− model is a unique model of SMA that shows several peripheral phenotypes present in human SMA patients. While a distinctive metabolic phenotype is seen in all Smn2B/− mice, the human population is more heterogeneous in its presentation of non-neuronal SMA symptoms. A portion of SMA patients present with higher instances of dyslipidemia, elevated markers of fatty acid oxidation defects, and microvesicular steatosis of the liver.14,34 Some patients also present with insulin resistance, impaired glucose tolerance, and metabolic syndrome.15,35,36 We have displayed that the liver may be an early target of SMA, with the potential to impact several other systems affected in the disease. The liver performs an important role in neuronal development, metabolism, and detoxification that should not be overlooked in the care and treatment of SMA patients. A holistic treatment strategy will provide the greatest outcome for patients.
With the increasing use of therapeutics, SMA patients’ lives are being extended and peripheral symptoms are becoming more apparent. It is crucial to understand the impact of individual tissues on the overall picture of the disease to treat patients as effectively as possible. Our work has provided an in-depth systematic characterization of the Smn2B/− mouse phenotype to allow for a better understanding of the connections between the systems affected in SMA and to guide future research with this model.
Footnotes
Acknowledgements
The authors thank the University of Ottawa Histopathology Core and the Eastern Ontario Regional Laboratory Association (EORLA). We are grateful to the Kothary laboratory for helpful discussions.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Muscular Dystrophy Association (USA) [grant number 963652 to R.K.]; the Canadian Institutes of Health Research [grant number PJT-186300 to R.K.]; the Fredrick Banting and Charles Best Canadian Institutes of Health Research Doctoral Research Award to A.R.; and the University of Ottawa Brain and Mind Institute CNMD STAR Awards to A.R., R.Y., and E.S.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author.