
Abstract
Inclusion body myositis (IBM) is an idiopathic muscle disorder primarily affecting adults above the age of 50. IBM is characterized by weakness in the knee extensor and deep finger flexor muscles due to muscle atrophy and fibroadipose replacement. Dynamometry and manual muscle testing are commonly used to assess patient muscle strength, while magnetic resonance imaging and electromyography studies identify the patterns of muscle atrophy and motor unit potentials. Although the underlying pathophysiological mechanisms of IBM are still unknown, common histopathological markers include rimmed vacuoles and inclusions. The immune system is also largely implicated in pathogenesis, as skeletal muscle in IBM overexpresses major histocompatibility complex I (MHC-I), and cluster of differentiation (CD) 8+ T-cells, and features endomysial inflammation. Antibodies to the cytosolic 5′-nucleotidase 1A (cN1A) protein have been associated with IBM but have low sensitivity and specificity. As many classic features of IBM present only in advanced stages of disease, there are substantial challenges to the diagnosis and monitoring of IBM progression in its early stages. Identifying early diagnostic biomarkers and new biomarker signatures associated with IBM disease progression is necessary for clinical trial readiness.
Keywords
Overview of inclusion body myositis (IBM)
Inclusion body myositis (IBM) is an idiopathic muscle disorder primarily affecting adults above the age of 50.1,2 Some studies have reported that up to 20% of patients with IBM have symptom onset at <50 years.3,4 Although initially characterized as an inflammatory myopathy, IBM has also been classified as a myodegenerative disorder.1,5–7 Inflammatory properties of IBM include the overexpression of cluster of differentiation (CD) 8 + cytotoxic T-cell infiltrates within skeletal muscle biopsies of patients through T-cell clonal expansion leading to antigen-driven inflammation.5–7 Degenerative features include the presence of rimmed vacuoles within muscle fibers that contain neurofilaments and aggregates of proteins associated with neurodegenerative diseases, such as β-amyloid, tau, and ubiquitin.5–7 However, not all muscle biopsies of individuals with IBM express these components, 5 and no highly sensitive diagnostic biomarker has been identified in all IBM cases, resulting in long delays between symptom onset and diagnosis, and misdiagnoses are common.2,6–11 Despite many investigated therapies, most patients with IBM are poorly responsive to immunosuppressive and corticosteroid treatments, unlike most other idiopathic inflammatory myopathies.7,10,12 Consequences of misdiagnosing IBM can be costly, both financially and in terms of patient burden, emphasizing the significance of a correct diagnosis at disease onset to minimize misdiagnosis and the risk of treatment-related morbidity.13–15
Clinical manifestations and prognosis
Since the initial description of IBM in 1971, 16 IBM has been recognized to cause insidious onset of painless, progressive focal limb weakness.6,9,12,17 Up to 2/3 of patients also develop dysphagia. 2 Initially, patients with IBM are diagnosed due to asymmetric weakness in the knee extensor muscles (quadriceps), deep finger flexor muscles (flexor digitorum profundus, flexor pollicis longus), and dysphagia due to cricopharyngeal muscle dysfunction (Figure 1).1,2,12,18,19 As the disease progresses, more widespread muscle involvement occurs in the ankle dorsiflexors, facial muscles, pelvic girdle, and neck flexors.1,20,21 Patients develop significant muscle atrophy of the quadriceps, forearm flexors, and diminished distal dorsal finger wrinkling. 22 Importantly, there is evidence of significant variation with regards to initial clinical presentation, progression, and accumulation of disability. 10 IBM leads to loss of ambulation, with 60–70% of patients requiring an assistive device to walk 7.5–10 years following onset of symptoms, and a wheelchair 13–15 years following onset of symptoms.12,17,23,24 IBM may slightly decrease life expectancy,25–27 with estimated life expectancy of 84.1 years in patients with IBM, versus 87.5 years in a control population matched for country, sex, and year of birth. 27 Given the slow progression and lack of clear diagnostic biomarkers, the average diagnostic delay is 5 years. 11
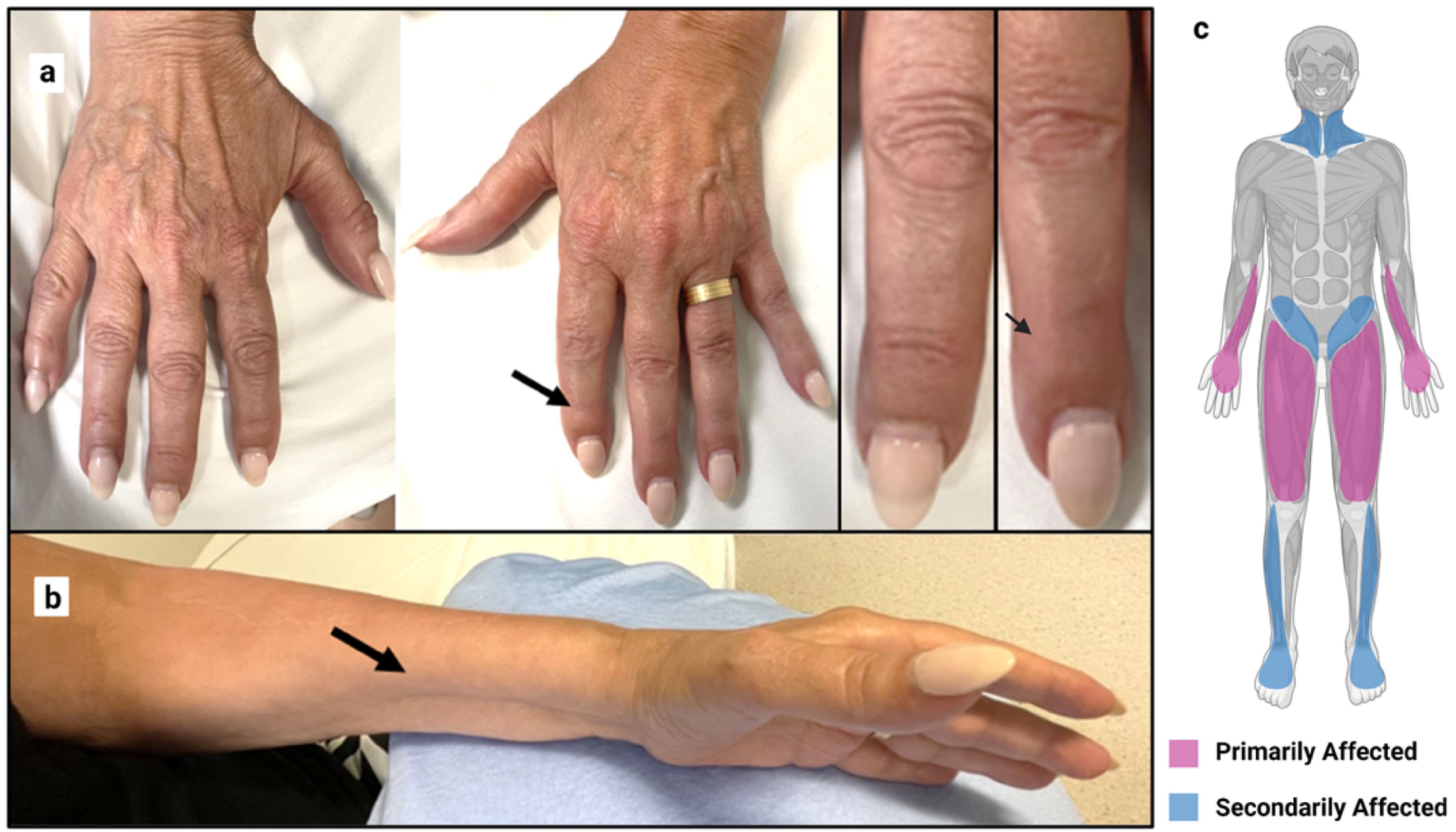
(a) IBM patient's hand demonstrating asymmetric diminished distal dorsal finger wrinkling (arrows) in the more affected hand, (b) IBM patient forearm demonstrating significant medial anterior forearm atrophy, (c) affected pattern of IBM patient disease progression, with muscle involvement beginning in the finger flexors (flexor digitorum profundus) and knee extensors (quadriceps) then spreading to the pharyngeal muscles, ankle dorsiflexors (tibialis anterior), and pelvic girdle.
Prevalence of IBM
Unlike many autoimmune diseases, IBM is more common in males than females (3:1 ratio). 2 A recent meta-analysis estimated the pooled prevalence of IBM is 46 patients per one million. 28 However, this prevalence is likely underestimated due to a high prevalence of patients with IBM misdiagnosed as having other autoimmune myopathies or genetic muscle diseases. 29 There is significant variability in prevalence (ranging from 1–71 cases per million 30 ), with the estimated prevalence of 5 per million in the Netherlands in 2000, 31 182 per million people over 50 years of age in the United States in 2010, 27 and 112 per million in Ireland in 2017. 32 IBM appears to be less prevalent in Korean, Mesoamerican Mestizos, and Middle Eastern populations compared to North American Caucasians. 33 The published IBM prevalence differences could be due to variations in diagnostic criteria (given establishment of diagnostic criteria after the first studies 34 ), or due to differences in geography, ethnicity, and genetic risk factors.
Diagnostic criteria for IBM
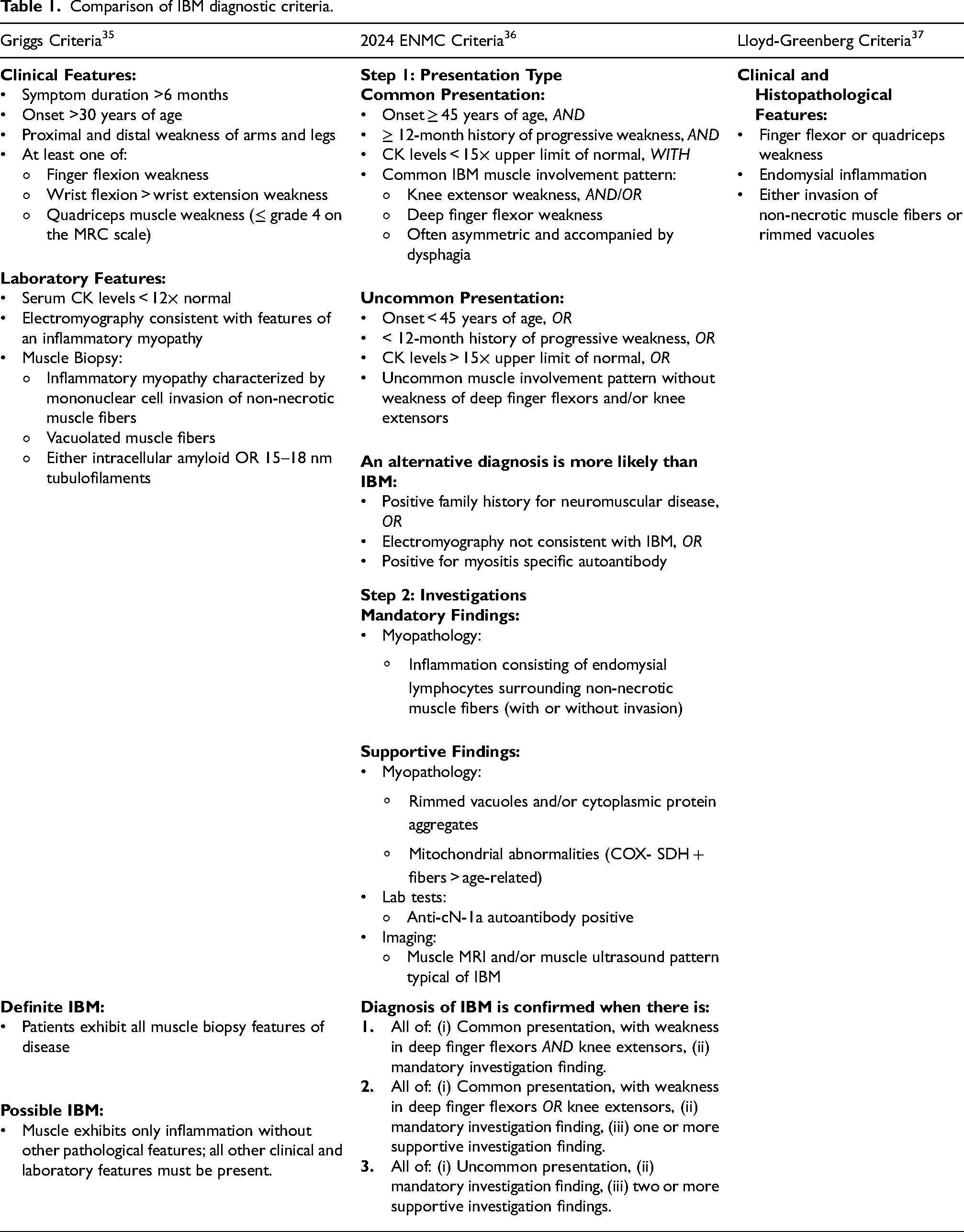
Several sets of diagnostic criteria have been established for IBM. Examples include the Griggs criteria, 35 the European Neuromuscular Centre (ENMC) 2024 criteria 36 which replace the ENMC 2013 criteria, 34 and the Lloyd-Greenberg data-derived criteria. 37 Summaries of diagnostic criteria are found in Table 1. Common features used to determine the diagnosis include questions about medical history (e.g., symptom duration, age, family history of disease), clinical evaluation with strength testing (knee extensors and finger flexors in particular), serum creatine kinase, myopathology, and electromyography. The specific tests recommended have evolved as our understanding of IBM has advanced. The minimum immunohistological stains recommended for state-of-the-art IBM diagnosis should include major histocompatibility complex class I (MHC-I) and II (MHC-II), CD68, CD8, CD45, membrane attack complex components c5b-9, and one of sequestesome 1 (p62), microtubule-associated protein 1A/1B-light chain 3 (LC3), and trans-activation response DNA binding protein 43 (TDP-43). 36 The ENMC 2024 criteria also introduce novel supportive diagnostic methods including serological testing for cytosolic 5′-nucleotidase-1A (anti-cN1A) antibodies, and muscle imaging using MRI and/or ultrasound. However, attempts to evaluate the performance of different diagnostic criteria have been complicated by the comprehensive and specialized nature of the investigations, with most patients lacking the full complement of tests required to assess all diagnostic categories.21,34 Similarly, applying machine learning techniques to identify features supportive of a diagnosis with IBM produced a list of features that were substantially influenced by the availability of the specific clinical test. 37 Clinical situations can arise where the assessment tools available do not permit a definitive diagnosis using established diagnostic criteria. For example, although myopathology investigations are generally considered mandatory, a muscle biopsy is not always feasible. In these situations, clinicians may opt to form a diagnosis based on clinical findings and supporting tests including muscle imaging and serological testing. 36 Establishing gold standard diagnostic criteria for IBM remains a challenge for the field. This is particularly true for early stage IBM, as established diagnostic criteria have reflected features typical of more advanced cases of IBM.
Comparison of IBM diagnostic criteria.
Clinical outcome measures
There are several clinically administered tests (questionnaires, strength testing) used to assess weakness and disease severity in IBM. The IBM Functional Rating Scale (FRS) questionnaire is used as a correlate of IBM disease severity from the patient perspective. 38 In the FRS, patients rate the level of difficulty for 10 tasks that are commonly affected in IBM on a scale of 0–4, with a lower overall score indicating decreased functionality in these tasks, and a lower score overall correlated with more progressed case of IBM. 38 The FRS has demonstrated reliability in describing disease severity, and correlates strongly to results obtained via a manual muscle test (MMT) and maximum isometric voluntary contraction testing (MIVCT), which assess muscle strength in the lower extremities of the body.38,39 However, the FRS is less reliable in describing upper extremity dysfunction, despite 6 of the 10 tasks heavily relying on finger function.38,40 Consequently, the IBM patient-reported outcome measures for upper extremity function (PRO-UEF) questionnaire was developed to specifically assess hand function in patients with IBM. 40 The PRO-UEF identifies 12 hand-specific tasks often affected by IBM and patients rate the level of difficulty completing these tasks on a scale of 0–4, with a lower score indicating more severe dysfunction. The PRO-UEF questionnaire correlates to measures of pinch and grip strength more strongly than the FRS. 40 Due to their strong correlations to objective functional tests, both of these questionnaires can serve as important clinical outcome measures to provide a better understanding of patient disease burden.
Manual muscle testing (MMT) with the Medical Research Council (MRC) score is commonly used during routine clinical visits to assess muscle function.17,41 MMT does not require specialized equipment, but has low inter- and intra-rater reliability when testing single muscle groups in myositis patients. 42 Additionally, MRC scores have limited sensitivity until dysfunction becomes severe, for example, an MRC grade of 4/5 may encompass up to 96% of the spectrum of potential muscle strength. 43 Isokinetic dynamometry is considered the gold standard method to assess patient strength through measuring the force it takes to resist against a patient's muscle action.44,45 Isokinetic dynamometry uses a machine to test the torque of a given muscle group at a fixed velocity by measuring the power required to resist the isolated muscle group's movement. 44 However, the isokinetic dynamometer is relatively expensive, time-consuming and requires significant amounts of clinical space. 45 Alternatively, handheld dynamometry (HHD) has demonstrated reliability and validity in quantifying muscle strength, and correlate with isokinetic dynamometer strength scores within the limits imposed by the strength of tester.45–47 Previous studies on patients with myositis other than IBM have demonstrated both inter- and intra-rater reliability when measuring muscle strength with HHD.48,49 Several studies on IBM have demonstrated good reliability for HHD to rapidly and cost-effectively assess muscle strength.48,50,51
For more global functional strength assessment, the timed up-and-go test, where patients are asked to rise from their seat, walk 3 m, turn around, and sit back down, has been used to test mobility in older adults and is a strong indicator of physical function in patients with IBM.52,53 A variety of timed and fixed-distance walking tests have also been used to indicate functional capacity in patients with IBM.52–54 However, these tests are less useful in assessing in patients with low endurance and those who are non-ambulatory.
Serological biomarkers
Anti-cytosolic 5′-nucleotidase 1A antibodies
Auto-antibodies play a major role in the immune system reaction and disease pathogenesis of patients with idiopathic inflammatory myositis (IIM).55,56 The anti-cN1A antibody is a 43 kDa protein associated with IBM,55,57 and the presence of anti-cN1A can support a diagnosis of IBM. 36 The cN1A antigen is a muscle-specific enzyme involved in metabolic regulation that catalyzes the hydrolysis of nucleotides and regulates nucleic acid degradation.58–60 In IBM, cN1A antigens can localize within rimmed vacuoles of the cell and may cause myonuclear breakdown where DNA degradation occurs. 59 Some studies have found that anti-cN1A antibodies are associated with worse clinical outcomes in IBM, such as more rapid deterioration of grip strength, 61 more severe motor lower extremity weakness, and more severe dysphagia symptoms. 62 However, others have demonstrated no significant difference in clinical outcomes of patients with IBM based on their anti-cN1A status.63–65 A recent study found that seropositivity for anti–cN1A-positive was associated with higher creatine kinase (CK) levels, but did not otherwise influence clinical or biopsy features or electrodiagnostic findings. 10 While anti-cN1A antibodies have moderate sensitivity and high specificity for IBM, their presence is not definitive for an IBM diagnosis.63,64,66,67 For example, elevated anti-cN1A antibodies have been identified in healthy controls, and patients with other IIMs such as dermatomyositis, and autoimmune disorders such as systemic lupus, although at significantly lower concentrations than those observed in IBM.55,66 Anti-cN1A antibodies have also been associated with a distinct phenotype of patients with a non-IBM IIM featuring a milder disease course. 68
Creatine kinase levels
CK is localized in the muscle sarcoplasm, myofibrils, and mitochondrial membrane, and can be found in higher levels in patient serum as a result of muscle breakdown. Serum CK levels are typically elevated in patients with IIM, with CKs up to 50× above the upper limit of normal (ULN). 1 However, CK levels are typically lower in IBM than most IIMs and range from normal to mildly elevated, 10–12× above ULN.1,17 Rarely, CK levels in IBM have been reported to be significantly elevated 20× above ULN, higher than ENMC criteria stating that CK levels should be less than 15× above ULN in definite cases of IBM.6,36
Imaging biomarkers
Muscle MRI can demonstrate focal areas of fibroadipose replacement, muscle atrophy, and edema in patients with IBM. 9 MRI scans of patients with IBM initially reveal asymmetric muscle atrophy and fibroadipose replacement in quadriceps and finger flexors, with more diffuse muscle involvement seen as the disease progresses (Figure 2(a)).69–71 In more advanced cases of IBM, the presence of an undulated fascia is often identified between fat-infiltrated and atrophic distal vastus lateralis and intermedius muscles, which represents complete muscle atrophy and fibroadipose replacement (Figure 2(b)). 71 Edema is characterized by a swelling of the skeletal muscle caused by increased fluid within the tissue, and is often found within the quadriceps muscles.72,73 IBM demonstrates the highest level of fibroadipose replacement, muscle atrophy, and edema in the anterior thigh (quadriceps) muscles, which can be useful in differentiating IBM from other IIMs. 72
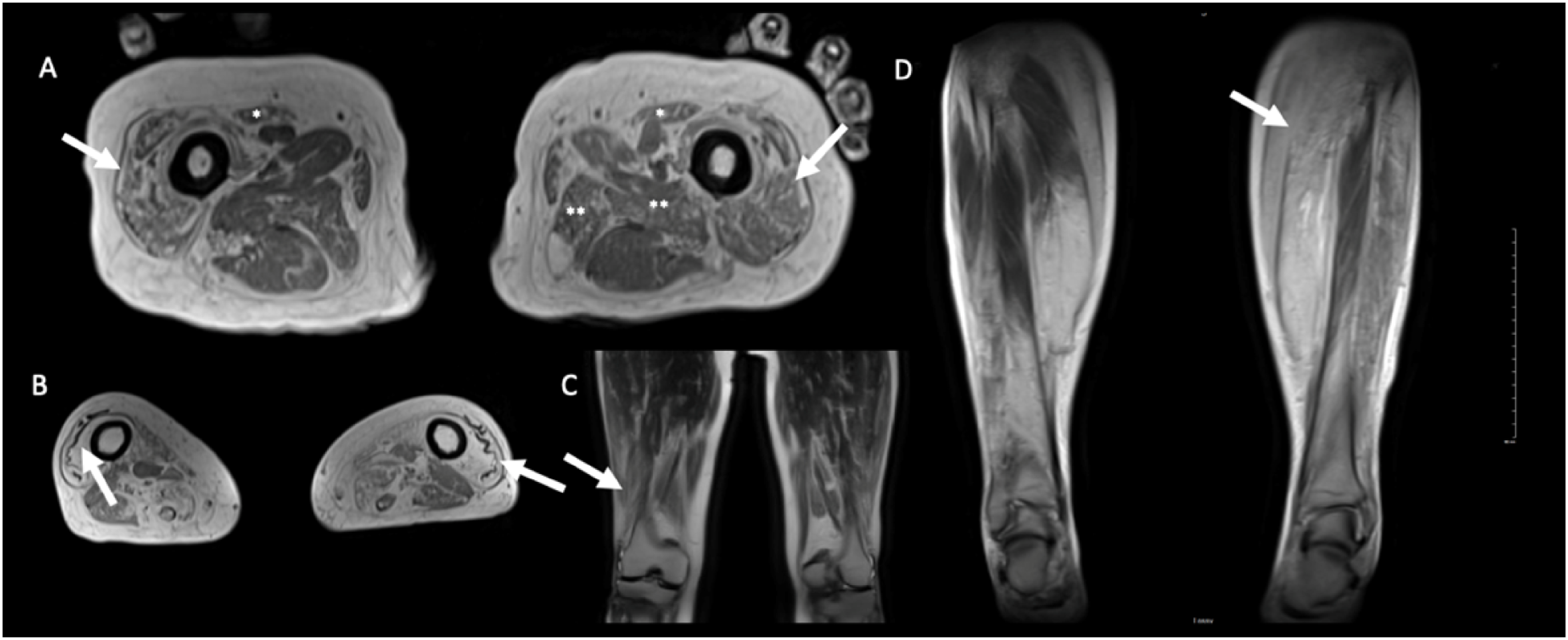
Axial VIBE in-phase images (5 mm thickness) of an IBM patient thigh demonstrating (a) bilateral moderate to severe atrophy and fibroadipose replacement of the quadriceps muscles (arrow), with variable atrophy and fibroadipose replacement in other muscles such as the sartorius (*) and left adductors (**), (b) the presence of a wave fascia (arrow) between fat-infiltrated and atrophic distal vastus lateralis and intermedius muscles, which present complete muscle atrophy and fibroadipose replacement. Coronal HASTE (5 nm thickness) images of IBM patient thigh demonstrating (c) a distal to proximal waste gradient, with more severe fibroadipose replacement more distally (arrow) and relative sparing toward the proximal aspects of the vastus lateralis and rectus femoris, (d) asymmetric involvement of the thighs, with the left thigh (arrow) being more affected than the right.
Electrodiagnostic studies
Electromyographic (EMG) studies are useful in distinguishing myopathies (with myopathic motor unit potentials [MUPs] small in amplitude and short in duration with early recruitment) from neuropathies (with MUPs that tend to be large in amplitude and long in duration with decreased recruitment).1,74 However, IBM can demonstrate a myopathic pattern, as well as a mixed or neurogenic MUP pattern in 32–56% of cases.74–78 Shorter MUPs (myopathic EMG pattern) in IBM correlates to increased quadriceps weakness and weaker strength score, while longer MUPs correlate to a more normal strength score. 79 While EMG can help inform the diagnostic pipeline, it cannot provide a diagnosis of IBM independent of other testing.8,76
Histopathological biomarkers
Despite the distinct clinical phenotype later in the disease course, histopathological analysis of the muscle tissue remains the gold standard for IBM diagnosis. EMG, ultrasound, and MRI can help identify the muscles most appropriate for biopsy. Histologically, IBM is characterized by variable degrees of endomysial inflammation with infiltration of non-necrotic myofibers by mononuclear cells (lymphocytes, macrophages), and the presence of rimmed vacuoles, and inclusions, in the context of chronic progressive dystrophy-like myopathic features (Figure 3). Active myofiber necrosis is a not a prominent feature, although regenerating fibers may be common. The inclusions originally described in IBM were the congophilic inclusions (with properties of amyloid) and tubulofilamentous inclusions identified by electron microscopy. 21 More recently, immunohistochemistry has identified various protein aggregate inclusions in the vacuolar myofibers in IBM, including TDP-43 and p62. Although p62 accumulation is common in inflammatory myopathies and is thought to be a general response to injury, 80 the pattern and location of p62 staining can be useful in differentiating subtypes of inflammatory myopathies, including IBM.81,82 Cytoplasmic bodies, although non-specific, are also commonly encountered in IBM.19,29,83 MHC-I and MHC-II are upregulated on immunostaining, with MHC-I having high sensitivity but low specificity for IBM, whereas a diffuse/patchy distribution of MHC-II can distinguish IBM from other IIMs with high specificity.21,36,84,85 Mitochondrial changes, including ragged-red fibers and an increased number of cytochrome c oxidase (COX) negative fibers, are also observed in a large number of patients with IBM. 86
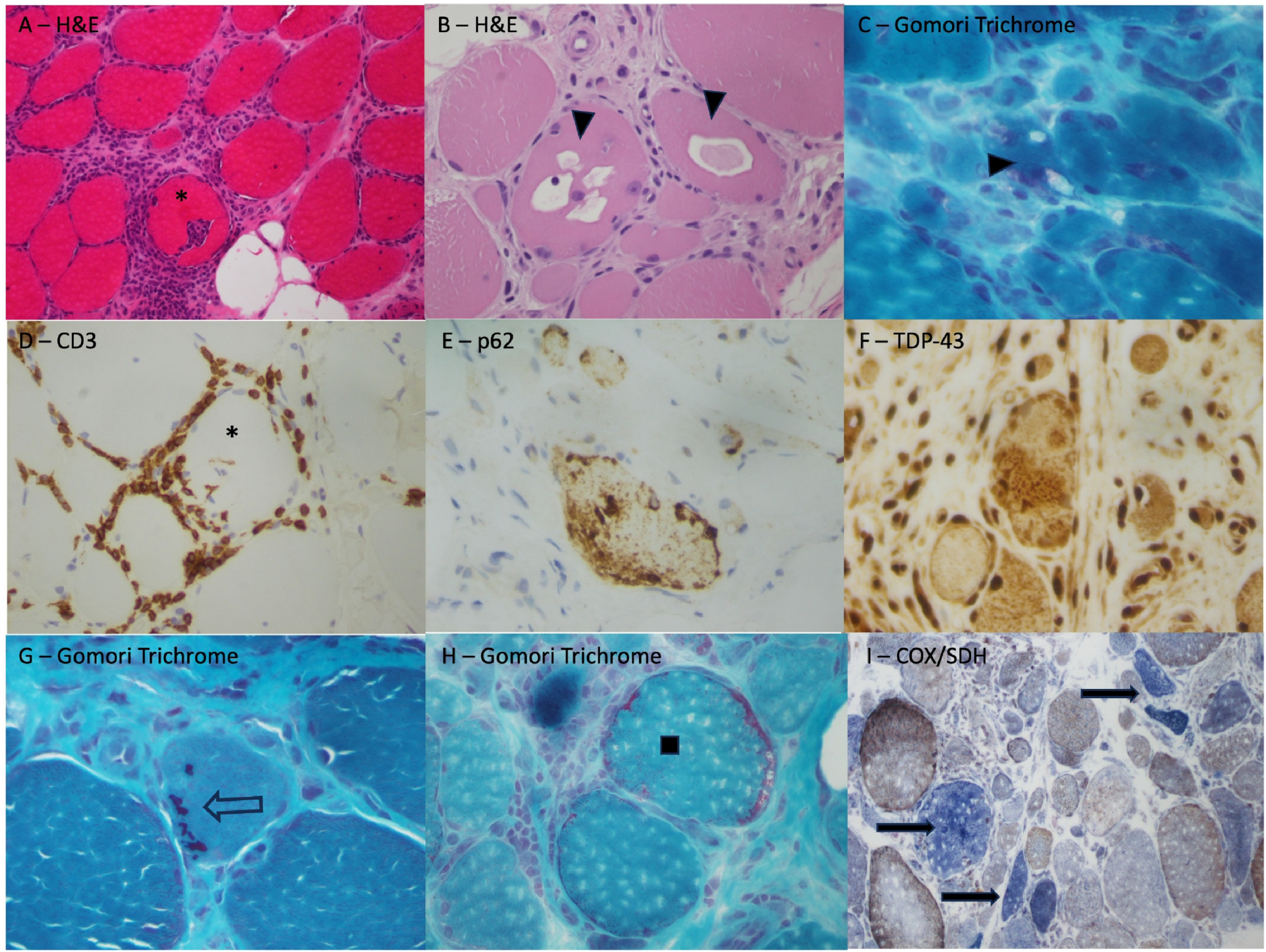
Muscle biopsy findings in patients with inclusion body myositis. Evidence of a chronic myopathic process with variable myofiber size (atrophy and hypertrophy), increased internalized nuclei, endomysial fibrosis and fibroadipose transformation/replacement with endomysial inflammation with invasion of intact myofibers [*] (A – H&E) and rimmed vacuoles [triangle] (B – H&E; C- Gomori trichrome). Immunohistochemistry for CD3 (D) shows a T-cell lymphocytic infiltrate surrounding and invading a myofiber*, and coarse protein aggregates can be identified with p62 (E) and TDP-43 (F) stains. Cytoplasmic bodies are common in IBM [open arrow] (F – Gomori trichrome). Mitochondrial changes may also be identified, including ragged red fibers (H – Gomori trichrome) and bright blue COX-negative fibers [closed arrows] (I - combined cytochrome oxidase [brown] and succinate dehydrogenase [blue] stains).
Rimmed vacuoles
Rimmed vacuoles (RV) are found in 66–90% of muscle biopsies from patients with IBM, and are considered a hallmark feature of the disease.7,10,18,87 RV contain protein aggregates that are thought to derive from myonuclei. 87 RV are characteristic of several other progressive myopathic diseases, such as oculopharyngeal muscular dystrophy and myofibrillary myopathies.2,88–90 RVs contain tubulofilaments, as well as autophagic markers like LC3 and p62.5,12,87,91 While protein aggregates are typically cleared by selective autophagy,91,92 these aggregates are not cleared and become contained within the rimmed vacuole in muscles of patients with IBM.87,92 The nuclear membrane proteins lamin A/C and emerin, along with myonuclear-associated histones, have been found in the rimmed vacuoles of patients with IBM.88,93 The presence of these aggregated proteins within the rimmed vacuoles points to myonuclear breakdown, which typically occurs as part of an autophagic process. 88
Intracellular congophilic deposits
Intravacuolar or extravacuolar aggregates of amyloid proteins, or congophilic materials, can often be found within IBM muscle fibers. 94 Cytoplasmic inclusions of beta-amyloid are visualized using Congo red and polarized light.92,95 The pathogenesis of these amyloid aggregates within skeletal muscle appears to be similar to that observed in neurons of Alzheimer's disease, although no correlation has been found between the genes implicated in Alzheimer's and IBM.19,96,97 β-amyloid 42 dimers, trimers, and tetramers were found to be increased in IBM patient muscle samples, which can become cytotoxic to the muscle fiber following aggregation. 98 This might be indicative of poor myoproteostasis, which leads to an accumulation of misfolded or aggregated proteins, eventually leading to myofiber destruction.98,99
Tubulofilamentous inclusions
Tubulofilamentous inclusions are often found in other neuromuscular disorders such as oculopharyngeal muscular dystrophy and congenital myopathy, and are a feature of IBM, as demonstrated by their presence in ENMC criteria categorizing definite clinico-histopathological cases of IBM.34,35,100–102 Tubulofilaments are indicative of muscle atrophy, but their exact pathophysiological mechanism in IBM disease pathology is unknown.
Mitochondrial dysfunction: cytochrome c oxidase deficient muscle fibers
Morphological alterations in IBM include abnormal mitochondrial proliferation (ragged-red fibers) and absence of histochemical COX activity. COX is a major part of the electron transport chain in cellular respiration and COX-deficient muscle fibers tend to contain reduced levels of normal mitochondrial deoxyribonucleic acid (mtDNA), along with multiple clonally expanded mtDNA deletions. 103 Although COX-deficient muscle fibers can be seen in normal aging (<1% of fibers), they are more common in muscle samples taken from patients with IBM (2–5% of fibers). 104 COX-deficient muscle fibers were among the most common findings in IBM muscle biopsies aside from upregulated MHC-I. 105 An overabundance of COX-negative, succinate dehydrogenase (SDH) positive fibers can support a diagnosis with IBM. 36 Also, individuals with a clinical diagnosis of IBM without mitochondrial defects may indicate another diagnosis such as dermatomyositis.81,106 This COX-deficiency was found to be associated with multiple mtDNA deletions within the affected muscle fibers. 104 One of the genes involved in mtDNA maintenance is the POLG gene, which encodes the mtDNA polymerase gamma protein. A variant or deletion of the POLG gene has demonstrated an induction of the accelerated aging phenotype in mice, which could result in more mtDNA mutations and eventually COX-deficiency. 107 Some variants of the POLG gene seem to be significantly more common in patients with IBM, and could be implicated in the disease pathogenesis. 86 COX-deficient muscle fibers are also more prone to atrophy than normal muscle fibers, indicating that mitochondrial instability may play a role in IBM disease severity. 108 Similarly, COX-deficiency is correlated to increased T-lymphocyte infiltration and muscle atrophy, demonstrating a potential link between mitochondrial deficiencies and the immunological pathogenesis of IBM.108,109 Cellular stress due to protein aggregation or chronic inflammation, can also lead to mitochondrial toxicity, which could activate the mitochondrial permeability transition pore (mPTP).110,111 mPTP can lead to dysregulated calcium concentrations through secretion of mitochondrial components, which is consistent in patients with IBM who tend to have elevated cytosolic and mitochondrial calcium concentrations and reduced expression of sarco/endoplasmic reticulum calcium ATPase protein 1. 112 An imbalance of intracellular calcium can trigger the formation of reactive oxygen species, which can lead to myofiber death. 110
TDP-43 protein localization
TDP-43 is a nuclear binding factor that aids in transcription regulation and RNA processing in the myonuclei.113,114 However, in patients with IBM, TDP-43 localizes within the sarcoplasm of skeletal muscle as opposed to within the myonuclei.115,116 This mechanism may be related to cell stress or inflammation, demonstrating that the inflammation pathway that is activated in IBM patient muscle may play a role in the mislocalization of TDP-43.114,117 Protein mislocalization may also relate to a general disruption in myoproteostasis, or disrupted autophagic processes, which have been implicated in rimmed vacuole production. Although the percentage of muscle fibers displaying sarcoplasmic aggregates of TDP-43 on a muscle biopsy varies significantly from 23%-78%,115,116 their presence has been demonstrated to be a highly specific immunohistochemical marker of IBM, and are a far more frequent finding than rimmed vacuoles and congophilic deposits.118,119 Mitochondria are also known targets of TDP-43, and pathological TDP-43 is highly colocalized with mitochondrial abnormalities in IBM, suggesting an interplay between TDP-43 accumulation and mitochondrial dysfunction in the pathogenesis of IBM. 120 These pathological changes such as RV, mitochondrial defects, and tubulofilamentous inclusions have been heavily associated with IBM muscle pathology, however, the impact of these pathological changes causing a rather distinct pattern of weakness in patients with IBM remains unexplained.
Immunological biomarkers
Muscle samples from patients with IBM also feature inflammation of the endomysial layer of non-necrotic skeletal muscle fibers via infiltration of mononuclear cells and other immune markers (Figure 4(a)).1,2,6 This infiltration may occur via increased expression of MHC-I on IBM patients’ non-necrotic muscle fibers.121,122 The exact antigen that triggers the upregulation of MHC-I is unknown, although several factors may contribute to the increased expression that is observed. Upregulation of MHC-I may occur initially as a result of pro-inflammatory signaling. 123 However, MHC-I overexpression also results in pro-inflammatory signaling, leading to a feedback loop between MHC-I and pro-inflammatory signaling. 124 MHC-I overexpression has also been implicated in vacuolization and autophagic protein accumulation in IBM, suggesting a link between the myodegenerative and inflammatory aspects of the disease. 125 An additional potential trigger of MHC-I overexpression is endoplasmic reticulum stress. 124 MHC-I molecules are typically found on the surface of all nucleated cells and are antigen-presenting to circulating immune cells, and when overexpressed, stimulate a T-cell immune response.126,127 In IBM muscle biopsies, the non-necrotic muscle fibers expressing MHC-I are invaded by cytotoxic CD8+ T-cells, demonstrating the formation of an MHC-I-CD8+ T-cell complex.95,122 The cytotoxic CD8+ T-cells observed in patients with IBM demonstrate the overexpression of the T-cell transcription factor T-bet, which is activated in response to an antigen and forms a clonally expanded set of cytotoxic lymphocytes. 126 Following cytotoxic lymphocyte formation, two distinct sets of CD8+ T-cells form.126,128 The first set involves non-senescent CD8+ T-cells, which overexpress the cell surface markers CD38 and human leukocyte antigen – DR isotype (HLA-DR), markers of T-cell activation that lead to constant formation of T-cells.126,128 The second set of cells are terminally differentiated memory CD8+ T-cells that overexpress the cell surface marker CD57, which represents a senescent cell.126,128 The overexpression of T-cells leads to the endomysial infiltration and inflammation observed in patients with IBM, and may play a role in IBM's primary pathology, since inflammation is associated with increased muscle atrophy.95,126,129 The event or stimulus that triggers the cascade of effects leading to T-cell overexpression in patients with IBM is still unknown, making it difficult to target in forming a treatment for IBM.

(a) Proposed pathway of immune system activation leading to muscle atrophy in IBM. A crucial unresolved question is whether the observed inflammation is primarily driven by autoimmune mechanisms, or a secondary response resulting from the degenerative pathway. (b) Summary of current diagnostic biochemical biomarkers used in IBM.
Current experimental therapies
Experimental pharmacological treatments
Although IBM muscle biopsies demonstrate inflammatory markers through CD8 + cytotoxic T-cell activation, immunosuppressive medications have not had long-term success in treating the disease.130–135 However, there have been therapeutic trials targeting the degenerative pathway of IBM. For example, arimoclomol is involved in prolonging the activation of heat shock factor 1, which is a transcription factor that controls the expression of heat shock proteins, effectively increasing their expression.136–138 Heat shock proteins are activated in response to proteomic damage, or proteotoxic stress, which can lead to protein misfolding and eventual aggregate formation. 139 Heat shock proteins are heavily involved in protein homeostasis through guidance of protein folding and disaggregation of proteins, 140 making them potential useful targets in IBM treatment. Arimoclomol has demonstrated the ability to decrease inclusion body formation, TDP-43 mislocalization, and overall cell death in IBM-like pathology-induced rat myocytes. 136
Non-pharmacological treatments
Although there are no established disease modifying therapies for IBM, moderate exercise may help maintain muscle strength and reduce inflammation levels4. Low-load blood flow restricted resistance training may decrease the rate of decline of muscle function in patients with IBM. 141 Resistance training has also been demonstrated in a rat model of IBM to increase mitochondria biogenesis, decrease beta-amyloid accumulation, and decreased rimmed vacuole presence and possibly improve mitochondrial function. 142 Further randomized control studies are needed on larger, well-characterized populations of patients with IBM to guide future studies on IBM management.
Some patients with IBM develop severe weight loss and malnutrition. However, there is limited and conflicting evidence regarding the benefit of medical and surgical interventions to improve dysphagia in IBM.143,144 Balloon dilation of the pharyngoesophageal segment is performed to decrease cricopharyngeal retraction and therefore keeps the upper esophageal sphincter open. 144 Although less invasive and inexpensive, two thirds of patients with IBM with balloon dilation of the pharyngoesophageal segment did not benefit from this procedure. 145 Cricopharyngeal myotomy has resulted in significant improvement of dysphagia symptoms in individuals with neurogenic dysphagia, 146 and oculopharyngeal muscular dystrophy. 147 More than 50% of patients with IBM who undergo myotomy report benefit.145,148 However, this procedure is invasive, and careful patient selection is important. Patients with IBM with failure of relaxation of the upper esophageal sphincter and preserved tongue and pharyngeal propulsion and laryngeal excursion are most likely to benefit. 149 Finally, botulinum toxin injections to the cricopharyngeus muscle and upper esophageal sphincter were found to improve symptomatic dysphagia and decrease aspiration. 150 Further studies are required regarding the optimal intervention to improve dysphagia in IBM. 151
Summary of IBM biomarker research
Due to the wide range of clinical and pathological symptoms found within patients with IBM, creating a gold standard set of diagnostic criteria remains difficult, especially early in the disease course. Both the pattern of muscle weakness and the rate of disease progression are highly variable, 2 and there is no unique biomarker that can be used to predict this progression or identify patients in early stages of the disease. 26 Serum biomarkers such as anti-cN1A antibodies and CK levels may contribute to the identification of IBM as a disease, but have not demonstrated the ability to aid in monitoring disease severity. 152 Specific muscle MRI and EMG patterns can help inform a diagnosis, but require context provided by other investigations. Myopathological biomarkers, such as rimmed vacuoles, congophilic aggregates, tubulofilamentous inclusions, TDP-43 aggregates, and COX-deficiency, while useful in identifying IBM cases, are not present in all patients, and repeat muscle biopsies in clinical trials are problematic.5,7 Immune biomarkers such as MHC II upregulation and CD8 + overexpression have also demonstrated specificity for IBM diagnosis, but no treatment targeting these markers have been successful in improving IBM symptoms. 2 Finally, current methods used in diagnosing IBM do not provide sufficient quantitative evidence to predict disease progression. 26 Expanded biomarker discovery is needed to provide unprecedented insight into biological pathways for therapeutic development, to enable earlier IBM diagnosis, and improved monitoring of the disease progression for clinical trials.
Footnotes
Abbreviations and acronyms
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ICS is supported by the Eric Poulin ALS Translational Research Fund. JWC is supported by a Department of Medicine University of Ottawa Clinical Research Chair and by grants from the Canadian Institutes of Health Research (INS-464765), Physician Services Incorporated(19–28), and Muscular Dystrophy Canada (932196). The funding agencies were not involved in the creation of this manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.