
Abstract
A novel semiautomated buffer exchange process workflow was developed to enable efficient early protein formulation screening. An antibody fragment protein, BMSdab, was used to demonstrate the workflow. The process afforded 60% to 80% cycle time and scientist time savings and significant material efficiencies. These efficiencies ultimately facilitated execution of this stability work earlier in the drug development process, allowing this tool to inform the developability of potential candidates for development from a formulation perspective. To overcome the key technical challenges, the protein solution was buffer-exchanged by centrifuge filtration into formulations for stability screening in a 96-well plate with an ultrafiltration membrane, leveraging automated liquid handling and acoustic volume measurements to allow several cycles of exchanges. The formulations were transferred into a vacuum manifold and sterile filtered into a rack holding 96 glass vials. The vials were sealed with a capmat of individual caps and placed in stability stations. Stability of the samples prepared by this process and by the standard process was demonstrated to be comparable. This process enabled screening a number of formulations of a protein at an early pharmaceutical development stage with a short sample preparation time.
Introduction
Use of laboratory automation in pharmaceutical formulation development workflows has grown substantially for both small molecules and large molecules (biologics), yielding productivity gains, conservation of precious material, and improved data quality and enabling impactful formulation development work to be conducted at earlier points in the pharmaceutical development process. Significant work has been done to facilitate solubility measurement1–7 and identification of solubility-enhancing formulations (typically low-viscosity liquid formulations of small molecules), and that work has been reviewed.8–10 Similar technology has been applied to higher viscosity liquids that are relevant to formulation development of oral liquid formulations as well as soft- and hard-gel capsules. 11 Automation of solubility determination has also been applied to polymer screening in the formulation development of solid dispersions. 12 Efforts to implement automation in forced degradation studies conducted to evaluate the solid-state stability of small-molecule drug substances in the presence of common excipients have been fruitful.13–16 This latter application is analogous to the protein formulation forced degradation studies presented in this work. The need for automated solutions in the biologics area has grown significantly with the rapid expansion of protein therapeutics in pharmaceutical development.
Recently, the number of biological drugs has been increasing. Large-molecule products such as proteins are typically more complex than small molecules due to more complex production methods. 17 An elementary step in the development of a new pharmaceutical drug candidate selected for clinical studies is the identification of a suitably stable formulation. For small-molecule drugs, formulation preparation primarily consists of adding and mixing drug substance with excipients. Unlike small-molecule drug substances, which consist only of a single component, the active ingredient, or drug molecule, a biologic drug substance is usually a solution that includes the biologic molecule (protein, antibody, nucleic acid, etc.), buffer species, water, and occasionally excipients. Biologics formulation preparation will have to deal with removing components other than the active molecules in a drug substance and replacing them with the components targeted by the formulator. To achieve this, a semipermeable membrane that can allow smaller buffer components to pass through but retain relatively large protein molecules is employed. Variations of this membrane process include centrifugation of ultrafiltration tubes, tangential flow filtration (TFF) with membrane cassettes, and dialysis, and these operations are usually very time-consuming and frequently not suitable for preparation of large numbers of formulations with small sample volumes. Many hurdles need to be overcome for automation of a buffer exchange process with small volumes and a large number of formulations. The major ones are to identify technology solutions to increase the number of formulations prepared simultaneously, to organize each of the solutions with unit operations, and to meet the requirement of sterile samples in packaging suitable for protein stability studies and similar to that needed at scale. To initiate formulation development, the formulation scientist is provided drug substance typically in a platform buffer such as phosphate-buffered saline (PBS), which is likely not optimum in terms of the chemical and physical stability of the protein. The formulation scientist is tasked with exploring the stability of the drug substance in solutions of various pH values, buffer types, buffer concentrations, ionic strengths/tonicities, tonicity modifier types, surfactants, antioxidants, and other stabilizers to support an optimum clinical shelf-life.
A general process of this approach is shown in Figure 1 . As noted by others, 18 from an automation perspective, this process is most conveniently separated into two activities: sample preparation and sample analysis. Analysis and analytical instruments are largely automated in an analytical or formulation laboratory, although some sample workup may be required.18,19 Sample preparation of a large number of formulations is resource intensive with respect to both personnel and drug substance, particularly at early stages in development. The result is a limited number of prototype formulations prepared, affording a minimal formulation design space and potentially leading to selection of a suboptimal formulation or at least a protracted development exercise. In addition, sample preparation techniques can vary widely between scientists, adding another level of complexity to interpreting the data and making comparisons.
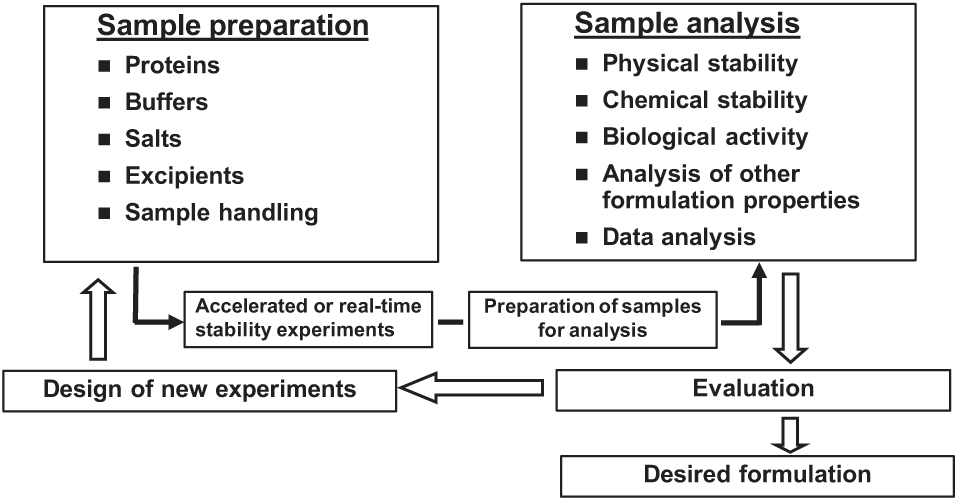
General protein stability and formulation screening process. The figure details the main areas of formulation development affected by automation—namely, sample preparation and sample analysis. 18
Automated approaches to stability sample preparation have been implemented.19–21 These involve diluting active solutions or powders (e.g., lyophiles) with formulation solutions to make prototype formulations. This approach is not preferred as it does not eliminate the original buffer components (in fact, the ratio of the original components to the drug substance may stay the same) and may affect the pH or other properties of the formulation. For proteins sensitive to changes in composition and pH, the results generated by this formulation preparation method may be confounding. In addition, the drug substance needs to be significantly concentrated or lyophilized before the prototype formulation preparation. These extra steps require additional processes to develop and may require large initial quantities of drug substance. They can also affect the stability of the molecule and can increase overall time-consuming operations in stability sample preparation.
The main aim of the present work was to create a semiautomated stability and formulation screening sample preparation process based on fully exchanging buffer components. This process should be suitable for generating a large number of formulations for stability testing with limited drug substance. This work will need to support the selection of a formulation for in vivo safety studies, including the earliest human trials. This imposes further constraints with regard to the data quality required to justify the decisions made. The present approach seeks to achieve efficiencies by standardizing on a parallel sample format (i.e., 96-well substrates) and introducing automated unit operations, while ensuring a high-quality sample output by incorporating sample pooling and redistribution and sterilization directly into glass vial containers. A case study screening the stability of an antibody fragment in formulations using the samples prepared by the workflow is presented.
Materials and Methods
Materials and Equipment
Protein BMSdab is an antibody fragment from the BMS portfolio and was provided as a solution in PBS at pH 7.2 by Biologics Process and Product Development, Bristol-Myers Squibb Co., Syracuse, NY. The excipients used were sodium citrate, citric acid, succinic acid, sodium chloride, histidine, sodium phosphate, and sucrose, all supplied by J. T. Baker (Avantor Performance Materials, Center Valley, PA). The water used was generated through reverse osmosis using Millipore Milli-Q UV Plus filtration systems (Millipore, Billerica, MA). These materials were selected as rational buffer components for the pH range under investigation—this process is amenable to incorporating other similar types of buffer components.
The equipment used for the semiautomated process was mainly small automated liquid handlers, a centrifuge, and automated solution volume measurement machine for plate wells.
A Bio-Tek Precision XS (Bio-Tek, Winooski, VT) with single and eight-pipette modes and an eight-probe Gilson 215 (Gilson, Middleton, WI) were automated liquid handlers used to add protein or buffer solutions. A modified model of the Apricot PP-550MS-XH (Apricot Designs, Covina, CA) 96-pipette automated liquid handler that is equipped with an automated filtration manifold (Pall Corporation, Port Washington, NY) was also used for liquid transfer and filtration.
An Allegra X-12R (Beckman Coulter, Indianapolis, IN) centrifuge was used for the ultrafiltration process of buffer exchange in conjunction with MultiScreen Ultracel-10 (Millipore) 96-well plates containing ultrafiltration membranes (also referred to as a molecular weight cutoff [MWCO] plate).
A VolumeCheck (BioMicroLab, Concord, CA) was employed for solution volume measurement in each of the 96 wells. A RoBo Rack-96 (Micronic LLC, Aston, PA) that holds 96 small glass vials and filter plates of 0.22 µm (Millipore) were used for sterile filtration of protein stability samples. An electric capmat sealer (Micronic LLC, Aston, PA) was also employed to seal the RoBo Rack-96.
Experimental Design
The semiautomated buffer exchange workflow uses a membrane separation process in a 96-well format, allowing the formulation scientist to leverage common automation tools for processing. Automated tools leveraging 96-well formats were identified for each of the unit operations to enable material-sparing approaches and reduce cycle time and scientist resources expended.
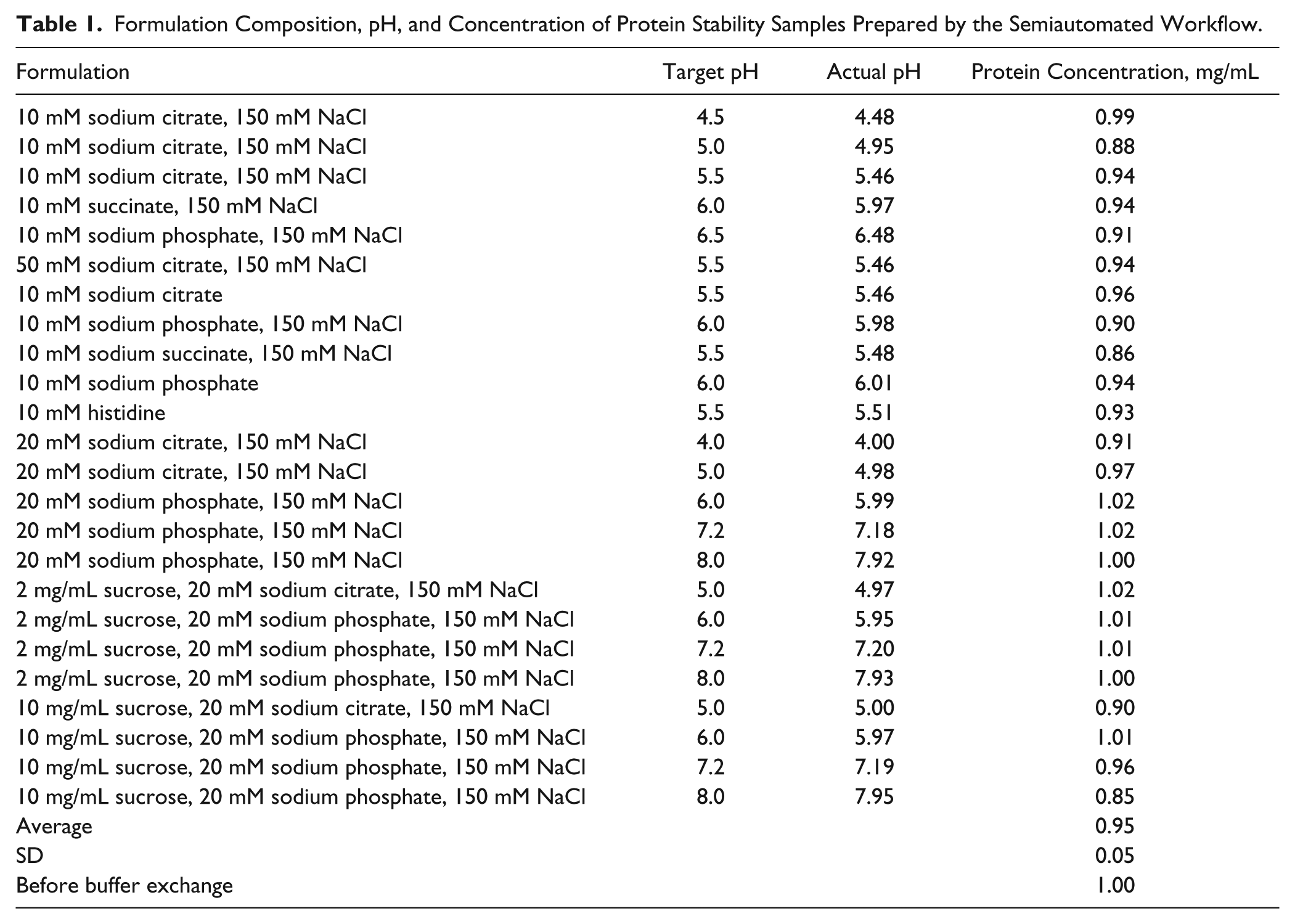
The experiment is designed to generate eight samples of identical composition and pH for use in formulation stability screening studies. These eight samples can then be distributed among the desired stress conditions and time points for a stability protocol while maintaining sterility of each sample independently. A 96-well plate format is used, with each column representing 8 wells of identical composition and pH. This allows for 12 compositions in each plate. Plates are easily run as one or two pair to occupy the rotors of a centrifuge. This typically means that 24 or 48 compositions with eight samples for each of the compositions are prepared in a single run. The BMSdab stability and formulation compositions were screened in two plates (24 formulations), and those formulations are given in Table 1 .
Formulation Composition, pH, and Concentration of Protein Stability Samples Prepared by the Semiautomated Workflow.
Semiautomated Sample Preparation and Analysis Process
Formulation buffers are prepared manually and are accurately transferred into a 12-channel plate with an automated liquid handler. The protein drug substance is then added into each of 12 buffers using a single or eight-pipette liquid handler. The actual amount of drug substance added may vary as the concentration of the stock is not standard but is typically 5 to 10 mg/mL. Sufficient protein drug substance is added so as to create solutions at 1 mg/mL at the target final volume ( Fig. 2 , step 1). The diluted drug substance is transferred into the ultrafiltration plate using a 96-pipette liquid handler. The ultrafiltration plate is then centrifuged at 3500 rpm at 5 °C to remove much of the original buffer solution while the protein therapeutic is retained and somewhat concentrated. Typically, two-thirds volume removal is targeted, and centrifugation time is adjusted accordingly ( Fig. 2 , step 2). After centrifugation, the plate is then placed in the VolumeCheck to determine the retentate solution volume in each well acoustically ( Fig. 2 , step 3). New buffer solutions are added by a liquid handler back to the target volume to replenish the sample volume ( Fig. 2 , step 4). This process is repeated until the buffer exchange efficiency is more than 99%. The number of cycles of buffer exchange required is determined by the targeted efficiency and is calculated based on the volume exchanged after each cycle. A spreadsheet is used for the automated calculation with volume value files generated by the VolumeCheck. The final step brings the wells back up to the target volume to once again achieve the target concentration of 1 mg/mL. After completion of the buffer exchange cycle, the 96-pipette liquid handler is used to transfer each column of samples from the ultrafiltration plate into a channel of the 12-channel plate to pool individual wells and achieve uniform concentration for a column ( Fig. 2 , step 5). The pooled solutions are mixed using several aspiration/dispense cycles, and then 500 µL of solution is transferred into each well of a 96-well sterile 0.22-µm filter plate (again, with columns of eight wells containing a given formulation) ( Fig. 2 , step 6). The samples are then vacuum filtered into the RoBo Rack-96, which contains 96 individual sterile glass vials ( Fig. 2 , step 7). Each of these steps leverages automated instruments. Last, the plate is then capped and sealed by the Micronic electric capmat sealer. Following this process, the plate contains 12 formulations with eight sealed, sterile 0.5-mL samples per formulation.
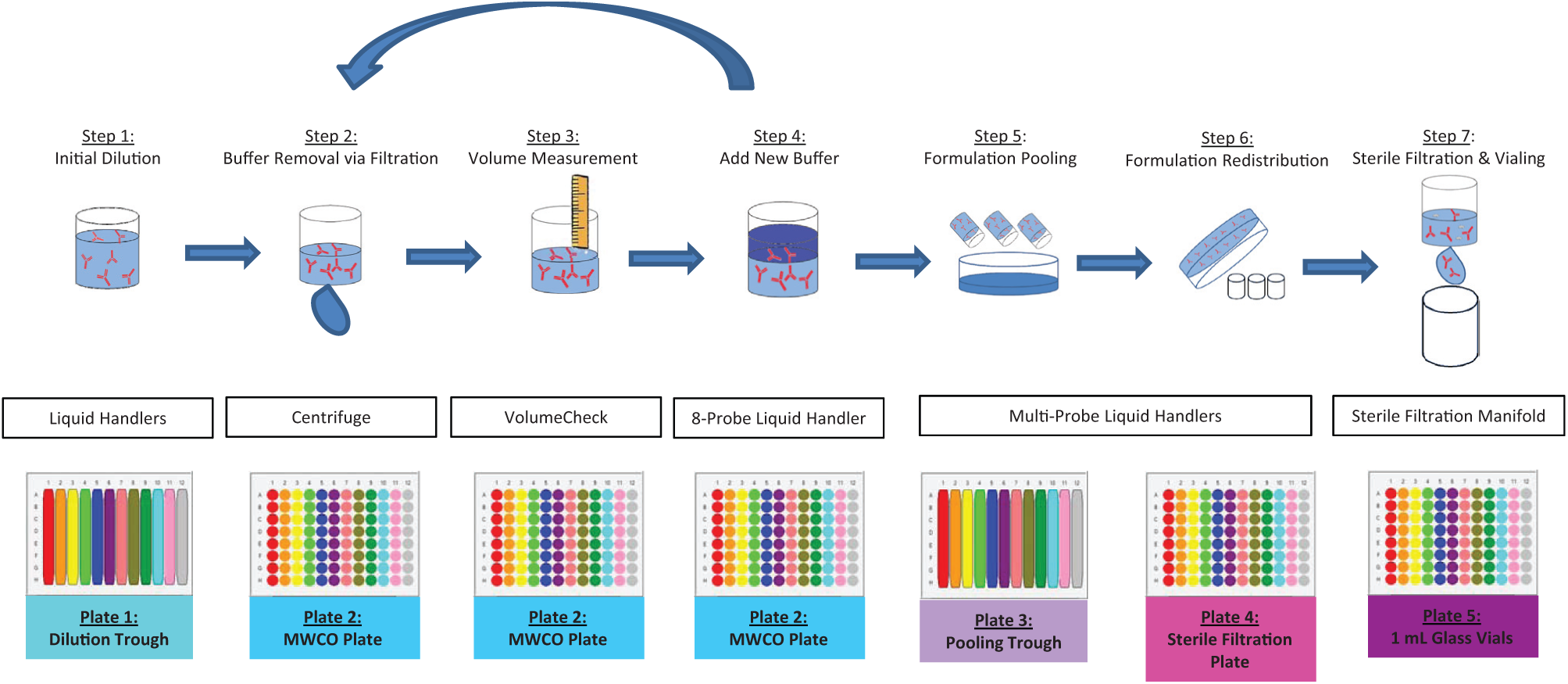
Buffer Exchange Sample Preparation Workflow. The seven key steps for sample preparation through buffer exchange are illustrated. Each step shows the unit operation occurring on each well, the instrumentation used, and the plate format. The protein is initially introduced to the process after necessary dilutions (step 1) traverse through the buffer exchange cycle (steps 2–4) several times until sufficient exchange has occurred. Finally, the sample quality is ensured through sample pooling and sterile filtration prior to vialing in steps 5 to 7. MWCO, molecular weight cutoff.
This process is designed to be expandable for a more automated process with additional capital investment of equipment such as a large liquid handler or robot arm with vial and plate grippers and suitable automation software for process and data collection/storage.
For formulation stability studies, the samples in sealed glass vials can be separated, placed in multiple holding plates, and placed into stability chambers maintained at different temperatures. The stability samples in glass vials are removed at predetermined time points and further processed for various analysis techniques. BMSdab stability samples were analyzed by size exclusion chromatography (SEC) after 4 weeks of storage at a stress condition of 30 °C. The autosampler for SEC was able to inject samples in a 96-well format. The stability samples were directly withdrawn from the glass vials. The SEC was selected for this study to directly measure aggregates and fragments of BMSdab stability samples. Compared with other high-throughput plate-based methods, this method provides direct reading of aggregate content and is also a standard routine method for assay of aggregation required by regulatory agencies for protein drug development.
SEC was performed on an AcQuity UPLC system (Waters Corporation, Milford, MA) consisting of a Binary Solvent Manager, a Sample Manager, a Column Manager, and a PDA detector. The separation of monomer, aggregates, and fragments was achieved using the GE Superdex 200 10/300 GL column (GE Healthcare, Princeton, NJ) and was detected by UV diode array detection at 214 nm with the reference wavelength set at 280 nm. An aliquot of 50 µL was injected. The column was maintained at ambient temperature. The mobile phase consisted of 200 mM sodium phosphate and 150 mM sodium chloride (pH 6.8) at a flow rate of 0.5 mL/min.
Results
The effectiveness of plate sealing was evaluated and minimal loss occurred under the extreme testing conditions. The average loss of solution weight in a capmat sealed plate of vials after 3 weeks at 50 °C was 3.36 ± 0.28 mg for a fill volume of 500 µL and 3.28 ± 0.21 mg for 200 µL. The loss of weight was similar between the 500-µL and 200-µL wells, which is consistent with the expectation of a surface evaporation phenomenon as the two volumes will present the same surface area facing air in the vial. Table 2 illustrates the percent change of solution weight for the 500-µL wells and 200-µL wells. All 500-µL wells showed no more than 0.78% of weight loss. For 200-µL wells, no more than 1.85% loss of weight was observed. To illustrate the buffer exchange efficiency, we measured pH and protein concentration in the plates prepared by the semiautomatic workflow ( Table 1 ). This is an indication of the quality of the buffer exchange experiment. Average protein concentration after the exchange process was 0.95 mg/mL (target concentration was 1.0 mg/mL) with a standard deviation of 0.05 and demonstrates that loss of protein was not significant. The final pH was within 0.1 pH unit of the target buffer pH for all buffer-exchanged samples. Figure 3 illustrates pH stability data for protein BMSdab, demonstrating consistency of results between the automated workflow and data generated through standard manual sample preparations as a control for the impact of the process.
Percent Solution Weight Change in Plate after 3 Weeks at 50 °C.
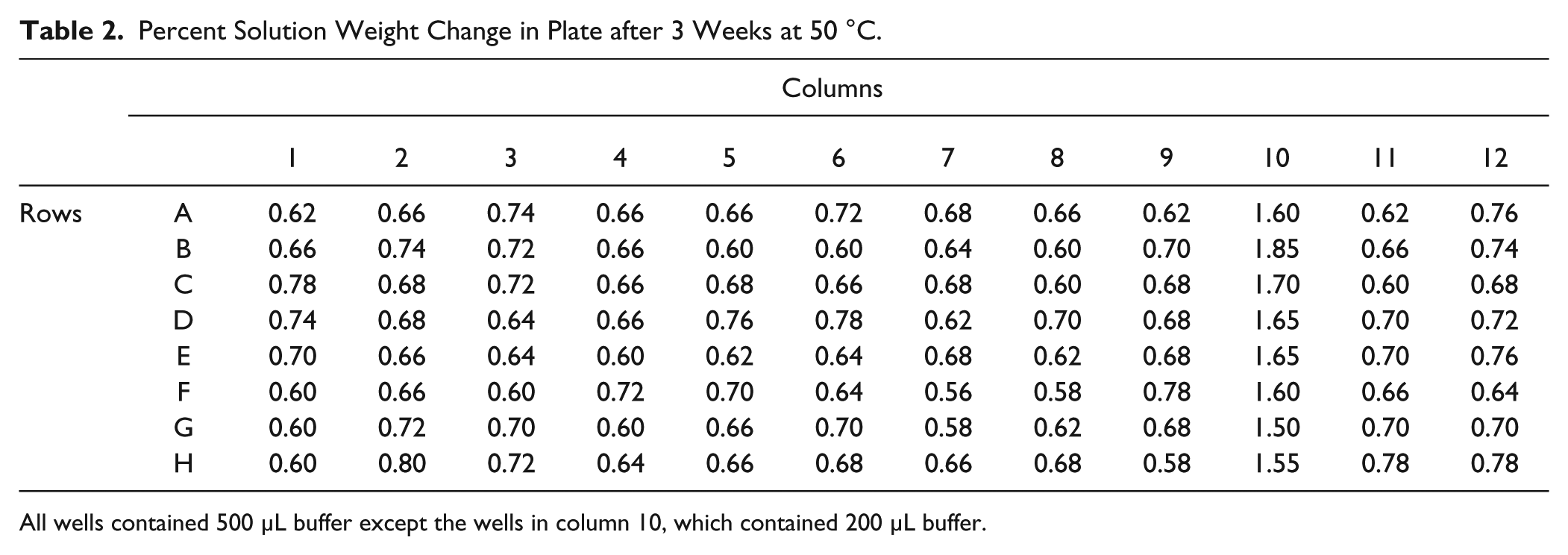
All wells contained 500 µL buffer except the wells in column 10, which contained 200 µL buffer.
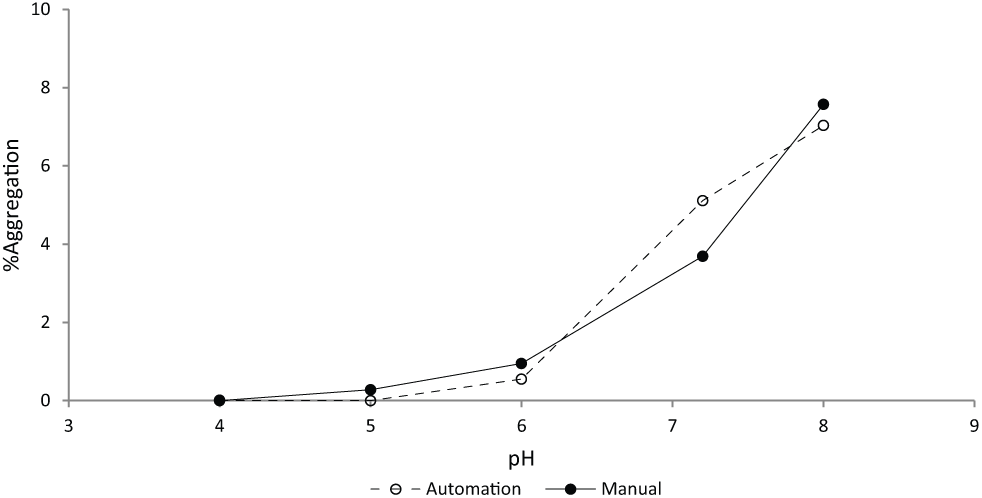
Comparison of physical stability of protein BMSdab observed in samples prepared using manual and semiautomated methods. A set of selected formulations was manually prepared and placed in the stability chambers. The manual preparation of formulations was completed using Millipore Amicon Ultra Ultracel-10K (Millipore, Billerica, MA) filter units for buffer exchange. All solution transfer manipulations were performed by manual pipette. The manually prepared stability samples were stored in 3-cc type I flint glass vials and sealed with 13-mm Daikyo D21-7S Flurotec-coated stoppers (West Pharmaceutical Services, Exton, PA) and an aluminum seal. Samples are stored at 30 °C for 4 weeks in 20 mM phosphate buffer, 150 mM NaCl.
The stability of 24 formulations of protein BMSdab was screened in a single run with 100 mg of drug substance using the samples prepared by the semiautomated workflow. The results are shown in Figure 4 . As presented in Figure 4a , protein aggregation increased at higher pH values, with sucrose having no observable effect on aggregate formation throughout the pH range studied. Fragmentation increased at lower pH values, with sucrose increasing stability at lower pH values. A pH range of 5.5 to 6 was optimal for minimizing both aggregation and fragmentation pathways. Figure 4b shows fragmentation and aggregation of protein BMSdab after 4 weeks at stressed conditions (30 °C) in various buffer types at pH 5.5 and 6. Protein BMSdab showed higher fragmentation rates in histidine buffer and lower fragmentation rates in phosphate buffer. Aggregation rates were relatively similar in citrate, succinate, and phosphate buffers but increased in histidine buffer. The presence of salt did not appear to affect aggregation substantially, as shown in Figure 4c , but did appear to adversely affect fragmentation, particularly at high pH. Higher buffer concentration adversely affected fragmentation at low pH but had less impact at other conditions ( Fig. 4d ). The sample preparation to support all of these studies was completed within 2 days as opposed to 2 weeks for sample preparation via the standard process. Based on these results, formulations containing protein BMSdab in phosphate buffer at pH 6, with sugars added for increased stability, were further evaluated, and a formulation suitable for preclinical and early clinical studies was developed.
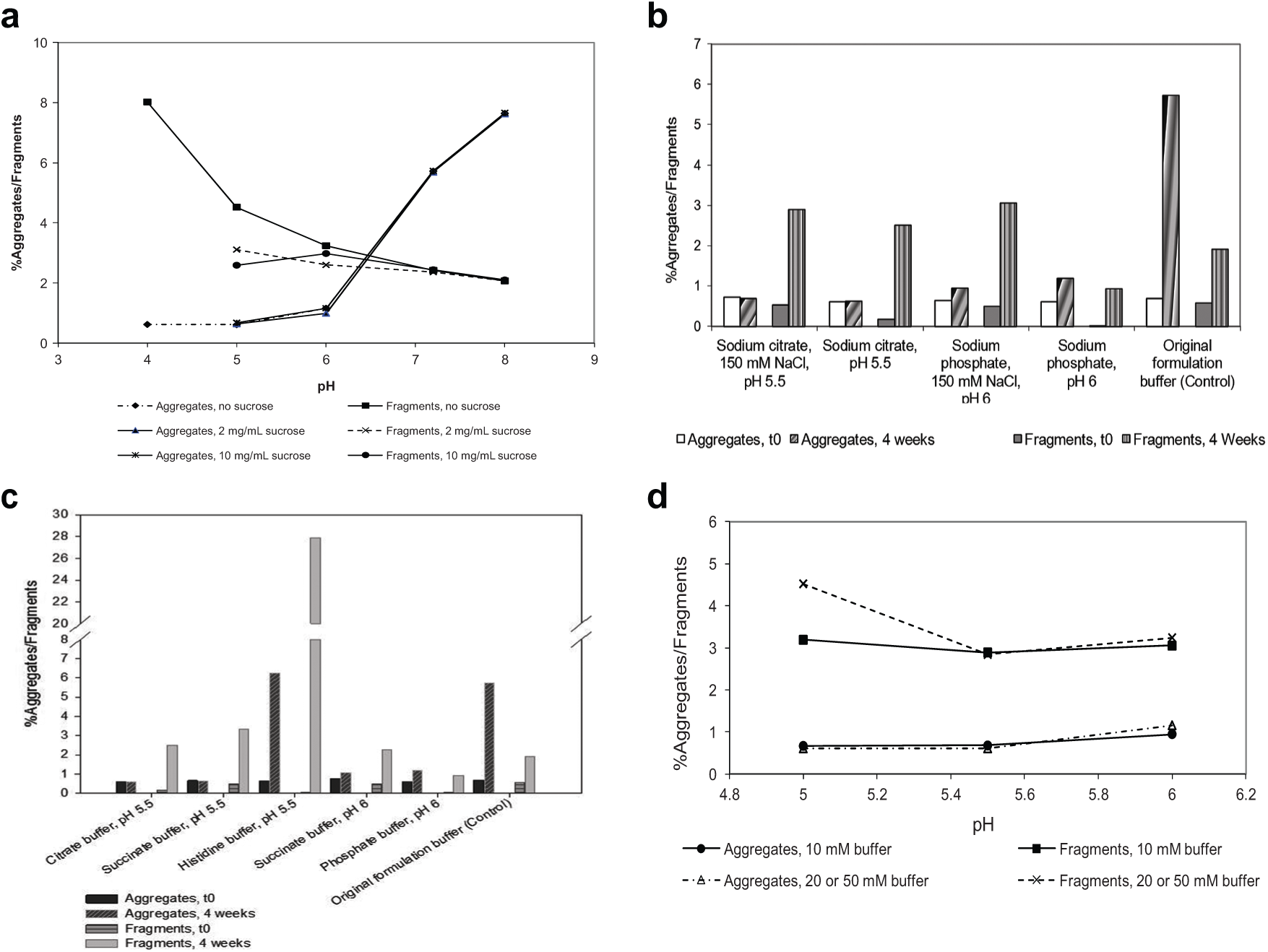
Stability results of protein BMSdab formulation screening after 4 weeks at 30 °C. Stability results from BMSdab formulation screening samples generated via semiautomated buffer exchange are reported. Samples were stressed at 30 °C for 4 weeks as measured by size exclusion chromatography–high-performance liquid chromatography and expressed as % aggregates and % fragments. Results are shown for (
Early in product development when this process was most relevant, the standard practice in our laboratories was based on dialysis exchange. The typical time required from buffer preparation to placing samples in stress conditions was 1 to 2 weeks for 12 to 24 formulations. The most time-consuming step was the exchange process. However, in addition to the exchange process itself, the logistics of staging each of the formulation buffers and samples in parallel for each of the process steps incurred a significant amount of time as well. Moving the process to (primarily) 96-well plates eliminated much of that logistical burden. The semiautomated process was executed in 2 days in comparison, affording a 60% to 80% savings. Another major benefit that was unforeseen is that due to the resource conservation of the method, this screening process could be conducted earlier in development to facilitate developability studies of candidates for development, helping to provide key information about the drug product development effort that may be required before investing further in a particular clone.
Discussion
There were several technical challenges in the development of this workflow. Several milliliters of sample per composition was desired, separated into single vials per time point/condition, driven by the consideration that sampling from a vial at each time point is effectively destructive. Microtiter format, automation-compatible buffer exchange plates were, however, only available on the microliter scale.
Accurately determining the amount of material retained on the filter membrane after each buffer exchange cycle was a challenge and was needed to ensure an appropriate amount of buffer was refilled into each well or column. Ensuring sterile samples were generated was another hurdle to be overcome. Last, this workflow was put together with specific hardware constraints, so the process was optimized around the tools available.
Due to these technical constraints, this workflow design traverses several format changes ( Fig. 2 ). Samples are dispensed into the buffer exchange plate as 96 discrete samples and then pooled together to render several individual samples equivalent. Last, the samples are separated again to accommodate filtration in 96-well plates and storage in separate stress conditions. Buffer exchange technology allowing this workflow to be accomplished in formats other than 96 wells might improve the efficiency of the workflow; however, the burden imposed is minimized due to the automation of the workflow. The VolumeCheck tool for measuring the volume of each well was essential to efficiently implementing the buffer exchange process. Sterile filtration at the conclusion of the buffer exchange cycles was a facile solution to our stability needs. Isolating large equipment in biological safety cabinets or sterilized enclosures is best avoided as this minimizes the utility of those instruments for other nonsterile purposes, and the current solution eliminates the need for a decontamination protocol. The RoBo Rack-96 plates resolved several issues for this application as well. Wells in typical 96-well plates are unable to be separated for exposure to various stress conditions or analysis at disparate times. It is also difficult to adequately seal these plates to maintain sterility, avoid contamination, and prevent evaporation at high temperatures during storage. In addition, plates are often made with plastic materials in contrast to the glass containers in which most biologics products are typically stored. Biologics are sensitive to surface interfaces and may behave differently in the two different container types.
It was noticed in this study that the amount of volume removed from each well by centrifuge differs due to fitting a rectangular substrate in a process governed by the physics of a circle. In our case, the plates were centrifuged in a portrait configuration, so the length of the axis of rotation varied down each column/formulation (which was horizontal when centrifuging). This causes the force to vary, and so the amount of material retained on the membrane varies (the highest force and swiftest filtration rate is along the outside, vertical edges). As the liquid handler used only has a single drive for each probe, the same volume must be added to each of the eight wells (which happen to all be of the same target formulation). The replacement volume for a column is equivalent to the volume required to fill the well that has least exchanged in each column (most volume retained—typically the center wells). Because the buffer exchange efficiency was different between wells in the same column, an average efficiency value for the column was used to determine the number of buffer exchange cycles. Applying vacuum on an ultrafiltration plate instead of centrifuge has shown more uniform exchange efficiency across a column or whole plate (unpublished data). Therefore, replacing centrifuge with vacuum-based ultrafiltration can applied to an expended and more automatic process.
Although this semiautomated workflow was established with specific hardware constraints, it could be a basis for further automation such as a more automated workflow or fully automated process by eliminating manual operations used in this semiautomated workflow. Particularly, the movement of plates can largely be automated within a deck by a large liquid handler system. Physically incorporating the VolumeCheck or a volume measurement sensor into the deck can also increase automation. The centrifuge process to remove original buffer can be automated by incorporating a suitable centrifuge to the deck. Alternatively, a pressure or vacuum system may be incorporated into the deck and applied to ultrafiltration plate to remove original buffer. Analytical instruments, such as pH meter, UV/Vis, and high-performance liquid chromatography, and stability chambers may be added into the large liquid handler system or reachable by an arm of the system to eliminate manual movement of plates for analysis. In addition, appropriate software development can also help to minimize or eliminate manual operations.
The utility of this approach is dependent on the drug substance being the only material retained on the filter membrane. Most other typical formulation components pass easily through the membrane, but components can tend to be retained because of size or surface activity such as polysorbate 20 or 80. It is important to ensure that the starting drug substance does not have any of these components to have confidence in the final composition generated. It is feasible to add these components from stock concentrates after buffer exchange in a well-designed study that might include them as factors.
Caveats/Considerations in Data Quality
Although protein formulation development is in many aspects analogous to that for small-molecule compounds, a number of features of a protein require special consideration. First, a protein can form secondary (alpha-helices, beta sheets, random coil), tertiary, and sometimes quaternary structures that are stabilized mainly by weak physical forces instead of covalent bonds, 21 and their integrity is part of the stability profile of the material. Stabilization of these structures depends on formulation but also protein concentration.
Second, the physical stability of proteins may be readily jeopardized. Moreover, like small molecules, there are also issues of chemical instability. Aggregation, fragmentation, oxidation, deamidation, and so on may occur readily under improper handling conditions. Conditions such as exposure to oxygen, light, heat, interfaces, and the presence of hydrophobic surfaces could occur in an automation process and cause protein stability problems. 22 For a protein, the significance and type of these stability problems may also be related to the various conditions to which the protein is exposed throughout the sample preparation process. Therefore, this workflow is not without trade-offs in terms of caveats for observing the absolute stability of the protein. Depending on the protein size and the viscosity of its solutions, the buffer exchange plates and protocol are likely to be effective at relatively low concentrations. While the basic stability trends at low drug substance concentrations can often be extrapolated to higher concentrations, and early high-throughput results may be useful in narrowing the number of formulations for further screening, it is important that stability at required protein concentration be confirmed upon availability of sufficient quantities of drug substance. Other potential sources of sample processing-related artifacts include the susceptibility of some proteins to agitation or phase interface-induced aggregation. For such proteins, the aspiration, dispensing, mixing, and vacuum filtration of the prototype formulations may contribute to generation of aggregates and affect data interpretation. The storage containers are selected to be analogous to what will be used on a large scale, but biologics can be sensitive to minute changes in extractables such as plasticizers, which may change from lot to lot and even more substantially from a small-scale stopper to a larger scale stopper.
In conclusion, a semiautomated sample preparation workflow for protein stability and formulation screening by eliminating >99% of the original buffer was successfully developed. The workflow does not require the sample to be concentrated to high levels for dilution or rendered solid by lyophilization or other process for aliquoting but uses a buffer exchange process to achieve the target formulations. Preparation of 24 unique formulation compositions with eight analysis samples for each composition for the early formulation screening of protein BMSdab was demonstrated with minimal drug substance (100 mg protein) and successfully led to discriminating between early stage formulation components for initial prototype formulations of the tested protein. The automation allowed formulation scientists to achieve the critical goals of using limited available drug substance, providing an avenue for standardizing screening procedures across scientists, and expanding the experimental space explored. The semiautomated process is also potentially expandable to a more automatic or fully automated process with additional capital investment for equipment and software.
Footnotes
Acknowledgements
We thank Drs. Lori Burton and Krishnaswamy Raghavan for assistance in proofreading this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.