
Abstract
Dissolution method transfer is a complicated yet common process in the pharmaceutical industry. With increased pharmaceutical product manufacturing and dissolution acceptance requirements, dissolution testing has become one of the most labor-intensive quality control testing methods. There is an increased trend for automation in dissolution testing, particularly for large pharmaceutical companies to reduce variability and increase personnel efficiency. There is no official guideline for dissolution testing method transfer from a manual, semi-automated, to automated dissolution tester. In this study, a manual multipoint dissolution testing procedure for an enteric-coated aspirin tablet was transferred effectively and reproducibly to a fully automated dissolution testing device, RoboDis II. Enteric-coated aspirin samples were used as a model formulation to assess the feasibility and accuracy of media pH change during continuous automated dissolution testing. Several RoboDis II parameters were evaluated to ensure the integrity and equivalency of dissolution method transfer from a manual dissolution tester. This current study provides a systematic outline for the transfer of the manual dissolution testing protocol to an automated dissolution tester. This study further supports that automated dissolution testers compliant with regulatory requirements and similar to manual dissolution testers facilitate method transfer.
Introduction
Historically, the assessment of drug release from oral solid dosage forms was based on tablet disintegration time. It was not until the 1960s that dissolution testing was introduced to assess drug release from dosage forms as a function of time. Dissolution testing is one of the most labor-intensive and time-consuming quality testing procedures involving a number of unit operations. In addition to the duration of the dissolution testing, there are numerous additional steps such as sample withdrawal, sample analysis, temperature measurements, media filling, media change, pH change, and vessel cleaning,1,2 often consuming twice the time of the testing procedure. As a result, pharmaceutical companies are now investing in more automated laboratory procedures, and a number of automated dissolution devices have been developed to increase the capacity while improving accuracy and reducing variability.
For immediate-release products, the entire dissolution testing procedure from sample introduction to complete release of drug from the dosage form is less than an hour. The setup of the dissolution apparatus for the next run and sample analysis is at least twice the time of the entire dissolution test. The poor time efficiency of manual dissolution testing for an immediate-release product necessitates the implementation of a more time-effective dissolution testing methodology. An automated system capable of a full-day testing routine, with efficient sampling and easier cleaning procedure, would be capable of conducting 10 dissolution test runs for an immediate-release product per day compared with an average of four test runs conducted by a well-qualified quality control scientist. In practical terms, an automated dissolution tester is capable of conducting 3000 dissolution runs for an immediate-release product in comparison to about 800 conducted on a manual dissolution tester under Good Manufacturing Practice conditions.
In addition, the increase in the number of modified and sustained-release drug formulations, which require much longer dissolution testing times (anywhere from 3–24 h), adds to quality control testing burden. Manual dissolution testing of modified-release products usually requires additional personnel, and often, a single dissolution test would be conducted by different individuals, resulting in higher variability and leading to repeated analysis in a number of cases. An out-of-specification result does require investigation and documentation, a major concern for the regulator and the industry in general.
The use of automation provides reproducible results with minimal personnel impact and electronically records data from all the test runs. Dissolution method transfer between laboratories is also a major concern. 1 These transfers become exceedingly challenging in the highly regulated pharmaceutical industry, where there is significant examination of all process aspects. 3 The objective of the current study was to evaluate a systematic method transfer from a manual to a fully automated dissolution tester, where the automated dissolution system was deemed the most efficient method compared with the currently validated manual technique. The selected product for method transfer was an enteric-coated aspirin tablet. Delayed-release products require dissolution media change and pH adjustments, providing additional challenges for the manual dissolution testing personnel (i.e., both time-consuming and shows magnified individual impact and variability). Aspirin is also an unstable drug at extreme pH conditions, making immediate sample analysis essential. The automated system was validated for the delayed-release product dissolution profiling of enteric-coated aspirin. After validation of the automated dissolution tester for this test product, reproducibility from one automated device to another was evaluated in different laboratory settings.
Materials and Methods
Materials
Enteric-coated 100-mg aspirin tablets were purchased from Bayer (Leverkusen, Germany). Aspirin, USP standard, was purchased from the United States Pharmacopeial Convention (USP, Rockville, MD). Potassium triphosphate was purchased from VWR (Leicestershire, UK). Hydrochloric acid was purchased from Merck (Darmstadt, Germany). Poroplast 10-µm filters were purchased from ERWEKA (Heusenstamm, Germany).
Preparation of Dissolution Media
In total, 0.1 M HCl was prepared by the dilution of hydrochloric acid 25% (w/w). Preparation of 0.20 M phosphate buffer (pH 12) was according to the USP. 4 Degassing of the media was conducted on a MediPrep 820 (ERWEKA) by heating the media to 38 °C, followed by vacuum degassing prior to dissolution testing for both the manual and automated dissolution testers. Dissolution media prepared according to the USP recommended manual method was compared with dissolution media prepared using a MediPrep 820 for verification of proper degassing procedure as a function of time. Dissolved oxygen in the prepared dissolution media was quantified using a GMH 3630 digital oxymeter (Greisinger Electronic, Regenstauf, Germany).
Dissolution Testing Parameters
A basket apparatus was used at a rotational speed of 100 rpm and maintained at 37 °C as described under USP chapter 711. The dissolution medium was 0.1 M HCl with a vessel fill volume of 750 mL (pH value of 1.2) for the initial 2 h. From 120 to 210 min, the dissolution medium was phosphate buffer (pH 6.8) by the addition of 250 mL 0.20 M tribasic sodium phosphate (total vessel fill of 1000 mL at a pH value of 6.80). Sampling was conducted at a single time point from the acidic dissolution media at 120 min, followed by sampling every 15 min after media pH change.
All standards were prepared in acidic and basic dissolution media. Standards were prepared prior to dissolution testing due to the sensitivity of aspirin in extreme pH media. Absorbance was measured on an Agilent 8453 Diode Array UV/VIS (Agilent, Santa Clara, CA), where the UV absorbance was determined at the isosbestic point of aspirin and salicylic acid, 278 nm and 267 nm for the acidic and basic media, respectively. The standard calibration curve was evaluated prior to every dissolution run; during all testing, the regression square exceeded 0.999. Control standards were measured after each cycle time point, and the relative standard deviation was less than 1%.
RoboDis II Vessel Fill
An integrated piston pump sequentially dispenses the dissolution media from the MediPrep 820 into the dissolution vessels. To validate the reproducibility of the fill volume for the acidic and the basic media at zero and 120 min, respectively, we conducted a volume fill verification study. The vessel fill was automatically conducted on a RoboDis II (ERWEKA) over 10 runs for the seven vessels, and the actual fill volume was compared with the programmed fill volume at the zero and 120-min time points.
System Performance Verification
For RoboDis II, since sampling is fully automated, the sampling lines are flushed between each time point. There are several volume options for the sample line flush to minimize contamination from the previous time point sample. 5 Since each vessel is equipped with an independent sampling line, cross-contamination between vessels is not possible. A 30-mL flush volume was selected based on a calculated fill volume of the sampling lines. Since the sampling lines are flushed prior to each sample time point, this may dilute the subsequent samples; therefore, a system performance verification test was conducted. In this study, dissolution testing was performed using an enteric-coated aspirin tablet in the RoboDis II following the same dissolution testing procedure described under Dissolution Testing Parameters. At each time point, samples were withdrawn automatically using the RoboDis II sampling port and with a manual sampling device from each vessel according to USP recommended protocol.
Run-to-Run Carryover
RoboDis II was programmed to wash the dissolution vessels following each dissolution run and to withdraw samples from each vessel prior to sample introduction to guarantee the absence of detectable drug levels. The current study was to determine the carryover effect from previous runs and the number of wash cycles required to completely remove drug residue. The automated system was set up for six runs; each dissolution run was followed by an indicated number of wash cycles, and the same dissolution testing protocol was repeated for subsequent runs (each test included a total of six tablets). The number of wash cycles was increased from one to three cycles for the evaluation of the suitable vessel cleaning protocol.
Equivalency Study between the Manual and Automated Tester
The study was conducted on three separate lots of enteric-coated aspirin over 3 consecutive days (n = 18 for each lot, six samples per day). The dissolution testing was conducted using manual dissolution tester ERWEKA DT 726 (ERWEKA), following the same protocol described earlier. The same lot was tested in parallel using an automated dissolution device, the RoboDis II. 6 The RoboDis II was programmed to withdraw samples simultaneously from all vessels, therefore reducing lag time between one vessel sample to the next. It was further programmed to electronically document each vessel temperature and pH prior to sample introduction, after media pH change, and at each sample time point, since most of the automated dissolution runs were conducted under minimal supervision; such documentation was crucial to assess validity of the data. At the termination of the dissolution run, the RoboDis II was programmed to measure the pH of each vessel to ensure that the pH was accurate during the dissolution run.
Equivalency Study between Automated Testers
Following the method transfer from the manual dissolution tester to the automated tester, the method transfer among automated dissolution testers was evaluated. In an automated tester, the absence of individual variability should result in a more reproducible interlaboratory method transfer. Three lots of enteric-coated aspirin tablets were tested over 3 consecutive days, with 10 runs per day and six samples per run on two separate RoboDis II systems in isolated laboratories. The RoboDis II systems were programmed and samples were placed in the sample door by different personnel.
Statistical Analysis
The mean and standard deviation (SD) of the results were presented. Data were analyzed using two-tailed Student t tests for the comparison of two groups and analysis of variance (ANOVA) for multiple groups where appropriate. Linear regression analyses were used to evaluate correlations and determine correlation coefficients.
Dissolution data were evaluated using the statistical formulas presented by the Food and Drug Administration. The similarity (f2) factor was calculated by using the following equation:
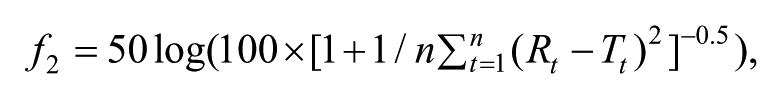
where Rt and Tt are the cumulative percentage of drug dissolved at each of the selected n time points of the reference and test product, respectively. The coefficient of variation at the earlier time points did not exceed 20% and at later time points were less than 10%, and only one time point was included after 85% release of the drug.
Results and Discussion
Dissolution Media Preparation
Since the basket apparatus is used for the current dissolution testing of enteric-coated tablets, dissolution media degassing is crucial to ensure reproducibility. The dissolution media degassed using a MediPrep 820 were compared with dissolution media manually prepared according to the USP recommended method (heating of dissolution media to 41 °C followed by media filtration through a 0.45-µm filter and gently stirring under vacuum) based on the quantification of the dissolved oxygen. The amount of oxygen quantified in dissolution media without any treatment and prepared by the USP manual method was 7.7 ± 0.3 and 3.50 ± 0.05 mg/L, respectively (n > 3). The dissolution media were prepared under vacuum for 1, 2, 5, and 10 min using a MediPrep 820, and the quantified oxygen was 3.9 ± 0.1, 3.68 ± 0.05, 3.4 ± 0.1, and 3.31 ± 0.1 mg/L, respectively. Since a 5- and 10-min MediPrep 820 treatment of dissolution media showed similar degassing efficiency in comparison to the USP recommended method, a 5-min MediPrep 820 treatment was used for the preparation of dissolution media for the manual and automated dissolution testers.
RoboDis II Vessel Fill
An integrated piston pump sequentially dispensed the dissolution media from the MediPrep 820 into the dissolution vessels at zero and 120 min with acidic and basic media, respectively. The vessel fill volume, verified over 10 runs for the seven vessels (six dissolution testing vessels and one reference vessel) (n = 70) to verify the reproducibility of the fill volume, was 749.77 ± 2.35 and 249.13 ± 1.79 for the acidic media and basic media vessel fill, respectively.
System Performance Verification
This study evaluated the suitability of the sample flush volume selected and sample line cleaning, as well as the risk of sample dilution within the sampling lines. The data, presented in Table 1 , show aspirin concentration presented as a percentage of label claim from each vessel from the automated and manual sampling device taken from the same vessel. The results indicate no significant difference between samples taken by automated sampling using the RoboDis II and manual device from the same vessel (ANOVA, p > 0.05). This study supports that there is no impact on sampling from sample line length or flush volume and no significant sample dilution within the sampling lines.
RoboDis II System Performance Verification Study for Enteric-Coated Aspirin Tablets.
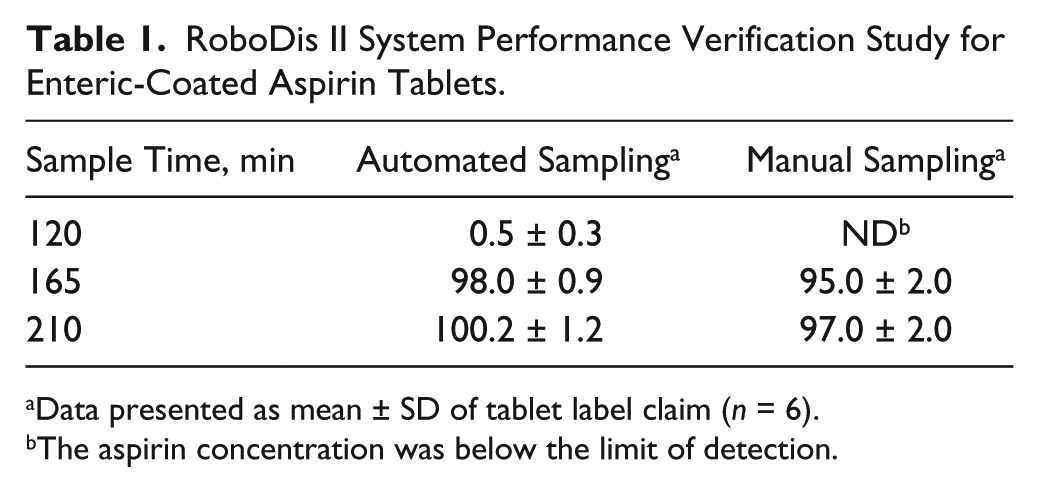
Data presented as mean ± SD of tablet label claim (n = 6).
The aspirin concentration was below the limit of detection.
Run-to-Run Carryover
The carryover study was conducted to determine the appropriate number of wash cycles following the dissolution run to minimize contamination of subsequent runs. Following each dissolution run of six enteric-coated aspirin tablets in individual vessels on the RoboDis II, washing of the vessel was conducted automatically with an indicated number of wash cycles using de-ionized water at room temperature. After the wash cycle(s), a new dissolution run was conducted using the same dissolution protocol to evaluate the efficiency of the vessel washing procedure. Prior to sample introduction into vessels, samples were withdrawn from each vessel as a blank sample to ensure the absence of residual aspirin in the vessels and the sampling system ( Table 2 ). The results for the acid media (0.1 M HCL) samples at the 120-min and 210-min time points (phosphate buffer, pH 6.8) were consistent after all wash cycles, with no sample carryover detected. The results support that adequate washing of the vessels and the sampling lines was achieved after a single wash cycle, and no residual aspirin was detected in the vessels or the sampling system. For all dissolution test runs conducted on the RoboDis II, the system was programmed to conduct two wash cycles to ensure vessel and sample system cleaning, and aspirin measured in the blank samples prior to sample introduction was lower than 1% of the tablet label claim.
Run-to-Run Carryover Study.
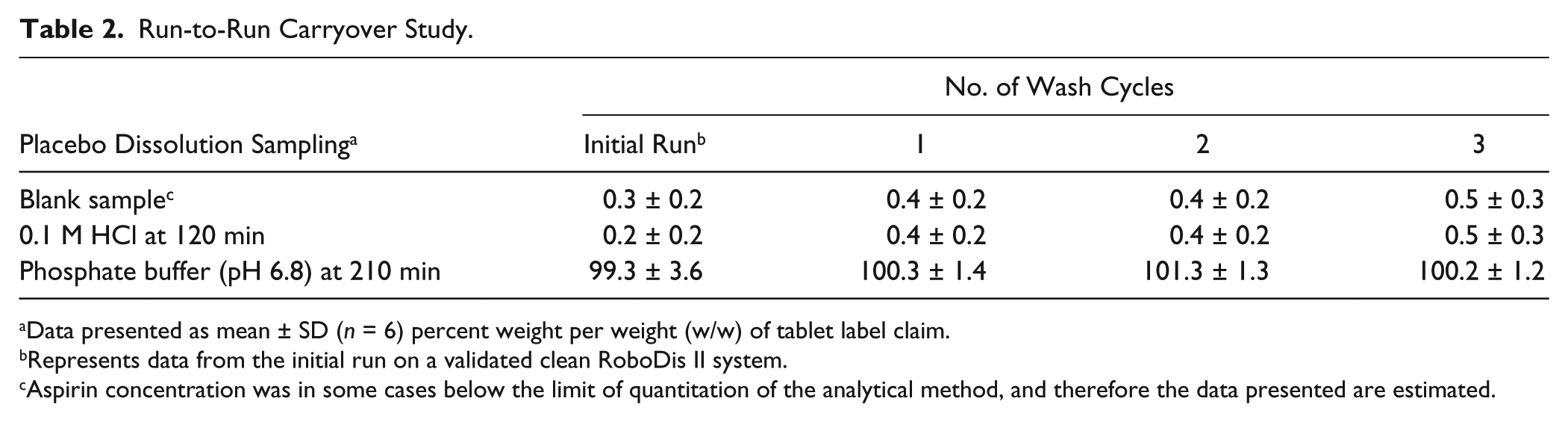
Data presented as mean ± SD (n = 6) percent weight per weight (w/w) of tablet label claim.
Represents data from the initial run on a validated clean RoboDis II system.
Aspirin concentration was in some cases below the limit of quantitation of the analytical method, and therefore the data presented are estimated.
Equivalency Study between Manual and Automated Tester
Six tablets from three separate lots were tested using both manual and automated dissolution devices following the same dissolution protocol over 3 consecutive days. The sample introduction and sampling from the vessels of the RoboDis II were performed simultaneously. The temperature was measured during each sampling time point in each vessel with the RoboDis II. The temperature of all vessels ranged from 36.8 to 37.2 °C during the entire testing procedure for all dissolution runs on the RoboDis II. The pH of the initial acid phase was monitored prior to test run. Subsequent to addition of basic media, the pH was monitored in all vessels. Since media pH change is automatically conducted on a RoboDis II, assessment of pH was essential for the validation of the pH change procedure, and the pH for all test runs was 6.79 ± 0.01. The dissolution data for both the manual and automated dissolution testing for the three lots are presented in Table 3 . The similarity factor is presented in Table 3 and indicates that the two dissolution techniques are similar for all lots tested, proposing that the automated method provides results similar to those obtained using the current manual validated method.
Method Transfer from Manual to Automated Dissolution Testing Equivalency Assessment.
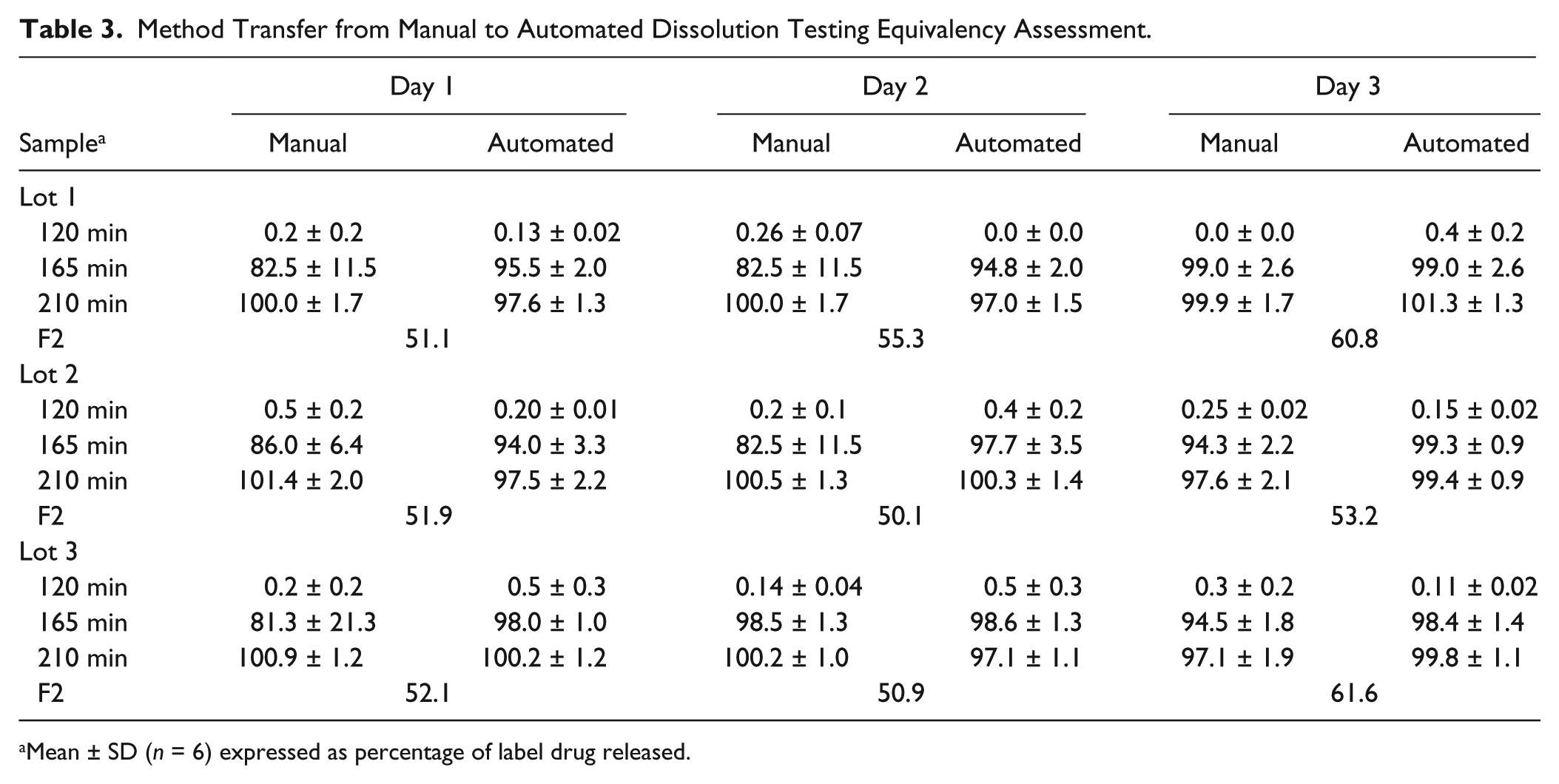
Mean ± SD (n = 6) expressed as percentage of label drug released.
Equivalency Study between Automated Testers
After method transfer from a manual dissolution protocol to a fully automated RoboDis II, the same method was transferred to another RoboDis II located in a different facility to evaluate ease of method transfer between automated testers. Three different lots were tested over 10 different runs during 2 consecutive days for a total of 60 runs on each device. Figure 1 provides the dissolution profile of enteric-coated aspirin tablets from the RoboDis II from two different laboratories. The coefficient of variation of all time points did not exceed 10% for the 60 runs. The study results were statistically evaluated by calculating the f2 similarity factor, comparing both dissolution profiles. The f2 value of 83.8 supports that both RoboDis II systems showed similar dissolution profiles. The ease of method transfer through software programming and the absence of individual variability facilitated the method transfer between both automated devices.
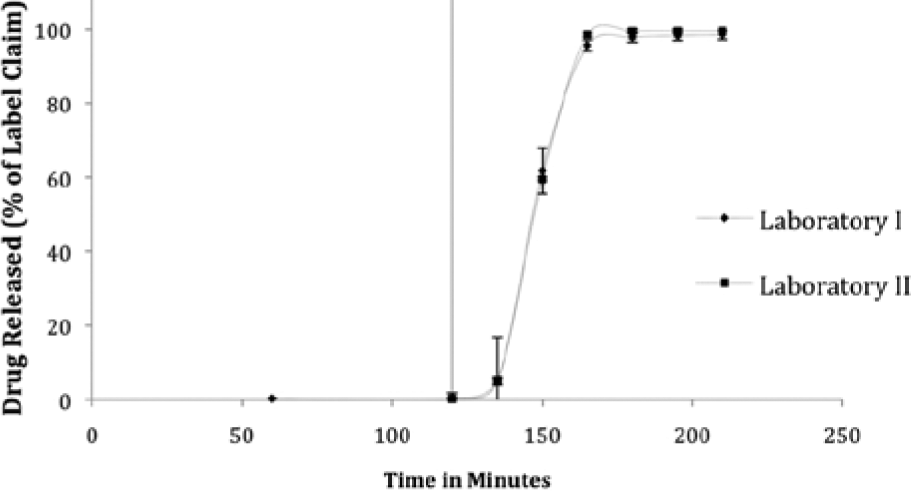
Dissolution profile of enteric-coated aspirin tablets tested by two RoboDis II devices located in different laboratories, programmed and operated by different technicians (n = 90).
In conclusion, there are a few automated dissolution testers on the market, and method transfer to a fully automated dissolution tester requires the assessment of the critical dissolution parameter attributes. Ideally, an automated tester should be fully USP compliant, showing the least deviation with regard to critical factors such as time gap between specimen introduction into vessels, position of sampling within dissolution vessel, and site of filtration, which should be conducted within the dissolution vessel to avoid removal of undissolved tablet fragments. RoboDis II used in this study is fully USP compliant; nonetheless, a full method transfer validation is essential. A number of criteria were evaluated for the proper dissolution method transfer from a manual to fully automated dissolution system. Transferring current methods from manual or semi-automated devices to fully automated devices requires planning and a number of systematic tests to validate the transfer and determine the suitable process parameters to eliminate sample carryover and sampling line contamination. The effective transfer is dependent on the product and validation of the dissolution method, and software setup requires knowledge and statistical equivalency verification. Once the method has been effectively transferred to an automated dissolution system, software programming facilitates method transfer among automated systems with minimal variation among units.
The use of automated dissolution testing devices is an added advantage to pharmaceutical companies, particularly with the increased number of required dissolution tests and the added requirements to narrow quality control specifications. Automated systems achieve high productivity and perform test runs continuously and automatically.
Footnotes
Acknowledgements
We thank ERWEKA for the equipment and facility access they provided for this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.