
Abstract
Current antiretroviral treatments target multiple pathways important for human immunodeficiency virus (HIV) multiplication, including viral entry, synthesis and integration of the DNA provirus, and the processing of viral polyprotein precursors. However, HIV is becoming increasingly resistant to these “combination therapies.” Recent findings show that inhibition of HIV Gag protein cleavage into its two structural proteins, matrix (MA) and capsid (CA), has a devastating effect on viral production, revealing a potential new target class for HIV treatment. Unlike the widely used HIV protease inhibitors, this new class of inhibitor would target the substrate, not the protease enzyme itself. This approach offers a distinct advantage in that inhibitors of MA/CA would only need to affect a subset of the Gag molecules to disable viral replication. To discover MA/CA-specific inhibitors, we constructed a modified MA/CA fusion peptide (MA/CAΔ) that contains the HIV protease (PR) cleavage site as well as a tetracysteine motif for fluorescent labeling. The HIV PR cleavage of MA/CAΔ can then be monitored via fluorescence polarization (FP). We have adapted this FP assay for high-throughput screening and validated it according to industry standards using a 384-well plate format. We have currently tested 24,000 compounds in this assay and here detail the screening methodology and the results of this screening campaign.
Introduction
The process of small-molecule drug discovery is long and expensive. 1 It begins with the identification of targets and early hits and then continues with an iterative application of medicinal chemistry and biological assays to optimize lead compounds. High-throughput screening (HTS) has become an integral part of hit identification, 2 so the HTS community continues to develop new techniques and assays to support the testing of new targets and novel approaches to existing targets. Fluorescence is widely used in HTS in vitro assays because of its simplicity, high signal to background, and because it can be readily used in homogeneous assays. 3
Much effort has been spent on identifying new anti–human immunodeficiency virus 1 (HIV-1) agents. While a variety of anti-HIV drugs have been approved and are in use, 4 the need for novel medicines to overcome resistance and reduce side effects remains. Current medications interfere with crucial receptors, enzymes, and corresponding functions of the HIV life cycle such as viral fusion with the cell, the reverse transcriptase, the integrase, or the protease. These drugs are very effective, but the appearance of resistant strains is a constant source of concern.5,6 Unfortunately, resistance to one drug often confers cross-resistance to another drug directed at the same target.7,8 A new anti-HIV target is the processing of the structural protein, Gag.9,10 This class of inhibitors is termed maturation inhibitors. Lee et al.11,12 have shown that a mutation at the MA/CAΔ cleavage site is strongly trans-dominant. A small amount of misprocessed structural protein, 15% to 20%, will completely ablate the infectivity of HIV-1, 13 suggesting that the MA/CAΔ processing site is an attractive target for HIV therapy.
In this article, we adapt a novel fluorescence polarization (FP) assay 14 to search for inhibitors that bind to the HIV-1 Gag protein and prevent its proteolysis. FP has been used extensively in protease assays,15,16 including assays of several viral proteases,17,18 but in all cases, the substrates were short peptides. In this assay, the 56-kDa MA/CAΔ fusion protein is labeled with fluorescein and used as the substrate, thus preserving a large portion of the Gag sequence as well as one of the protease cleavage sites. The MA/CAΔ polypeptide is modified by a tetracysteine motif (CCPGCC) to allow attachment of a fluorecein label (Fluorecein Arsenical Hairpin [FlAsH] reagent) and to create a greater size difference between the substrate (56 kDa) and proteolytic product (15 kDa).
Materials and Methods
Materials
The MA/CAΔ protein substrate and its DNA construct have been described.12,14 Purified HIV-1 protease (PR) was purified from Escherichia coli inclusion bodies as described previously. 19 FlAsH tag (Lumino Green in-Cell Labeling Kit, #12589-057) was purchased from Invitrogen (Grand Island, NY). Dithiothreitol (DTT) (43815), EDTA (E5391), HEPES (H3375), and HIV Protease Substrate 1 (H6660) were obtained from Sigma-Aldrich (St. Louis, MO). DMSO (2951) was purchased from EMD (Waltham, MA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) Criterion TGX Stain-Free gel (5678124) and SDS-PAGE loading buffer were obtained from Bio-Rad (Hercules, CA). The 384-well microplates (3575) were from Corning (Tewksbury, MA).
Chemical Library
The library was selected from 170,000 compounds that have passed more than one of the four Lipinski drug-like properties,20,21 have less than 10 rotatable bonds, and have passed all of the four-dozen reactivity filters proposed by Hann et al. 22 and Pearce et al. 23 The 24,000 selected compounds were dissolved in DMSO and shaken overnight at 300 rpm on a G10 Gyrotary (New Brunswick Scientific, Edison, NJ). Each orthogonal pooled library (OPL) was constructed as described earlier.24,25 Briefly, we used a Janus Pipetting Workstation (PerkinElmer, Waltham, MA) to transfer 10 µL of a 10-mM compound from ten 96-well source plates to five 384-well plates. Each well from the source plate was delivered twice and recombined with wells from the other nine plates. The pools in the 384-well plates contained 10 compounds at 1 µM in 100 µL. Screening data from OPL were de-convoluted using the10XYZ OpenHTS module (CeuticalSoft, Inc., Hudson, NY). 26
Liquid-Handling Robotics
Assay reactions were prepared in 384-well plates. Compounds were added to the wells with a 384-tip liquid-handling robot (NanoScreen NSX 1536) equipped with low-volume tips (#30 ulnc; NanoScreen, Charleston, SC). Substrates and enzymes were added with a MultiDrop Combi (5840400; Thermo Fisher, Waltham, MA) with a small cassette head (24073290).
Fluorescent Polarization Assay
Fluorescent labeling of the substrate: Purified 2.3 µM MA/CAΔ was incubated with 0.2 µM FlAsH in 50 mM HEPES at pH 6.8 with 1 mM DTT at room temperature for 1 h. Then, 13 µL of a 2.3-µM labeled substrate was added to assay plates containing either 3 µL of 10 µM of inhibitors (columns 3–22) or 1% DMSO (columns 1–2 and 23–24). After incubation for 15 min at room temperature, HIV-1 protease was added to columns 1 to 22. Heat-inactivated protease was added to columns 23 to 24. We have also found that saquinavir at 50 nM produced the same inhibition as heat-inactivated protease (data not shown). The final proteolysis reaction contained 1 µM MA/CAΔ 1 µM inhibitors and 100 nM protease.
After a 2-h proteolysis at 37 °C, FP values were measured, with excitation at 485 nm and emission at 530 nm using an Aquest GT Microplate Reader (Molecular Devices, Sunnyvale, CA). The data were analyzed using software from ScreenAble Solutions (Chapel Hill, NC).
Fluorescent Assay of HIV Protease Substrate 1
The assay was carried out as described above for the FP assay, except HIV protease substrate 1 instead of MA/CAΔ was used. The fluorescence was measured with excitation at 340 nm and emission at 490 nm using an EnVision Microplate Reader (PerkinElmer).
Gel Assay
A HIV-1 PR assay was performed as described for the FP assay, except that 2 µM FlAsH was used during the labeling procedure. After 2 h, 20 µL of the reaction was stopped with 4× SDS-PAGE sample buffer, analyzed by SDS-PAGE, and visualized using a gel scanner (Bio-Rad) with excitation at 488 nm and emission at 530 nm.
Data Analysis
Percent inhibition values for the FP assay and fluorescent assay values from each plate were analyzed using ScreenAble software. The Z′ for each plate 27 was determined using columns 1 and 2 as positive controls and 23 and 24 as negative controls. IC50 values were calculated with 95% confidence intervals based on a sigmoidal dose-response curve using a four- or three-parameter logistic equation featured in the GraphPad software (GraphPad Software, La Jolla, CA). The GraphPad Prism 5.0 program produced the graphic representations in the figures.
Results and Discussion
Primary Single-Point FP Assay
The goal of our screen using the MA/CAΔ substrate is to discover small molecules that bind to the HIV-1 protease-processing site. These processing site inhibitors will not inhibit the HIV-1 protease enzyme directly but bind to the substrate to prevent its proper proteolysis for the production of structural proteins needed for viral replication. The MA/CAΔ substrate is modified with a GST tag added to the N-terminus of MA, a truncation of the C-terminus of CA, and the addition of a tetracysteine motif in the N-terminal domain of CA ( Fig. 1A ). When the FlAsH reagent binds, it makes a fluorescently labeled MA/CAΔ substrate. When HIV-1 protease cleaves this substrate (56 kDa), two products are formed–a fluorescent labeled 15 kDa and an unlabeled 40-kDa peptide ( Fig. 1B ). In the FP assay, the 56-kDa substrate is large so that it does not tumble in solution and the FP value (mP) will be high, while the cleaved product of 15 kDa tumbles freely in solution, resulting in a low mP value. Figure 2 is a flowchart illustrating the assay process for screening the LCGC (Lankenau Institute for Medical Research Chemical Genomic Library) pooled library against the HIV-1 protease. The results of the HTS validation of this assay are shown in Figure 3 . The validation was performed by testing duplicate full-plate assays of high and low signal on 3 consecutive days. 28 The assay range and the Z′ of each plate are shown. All the Z′s are above 0.85.
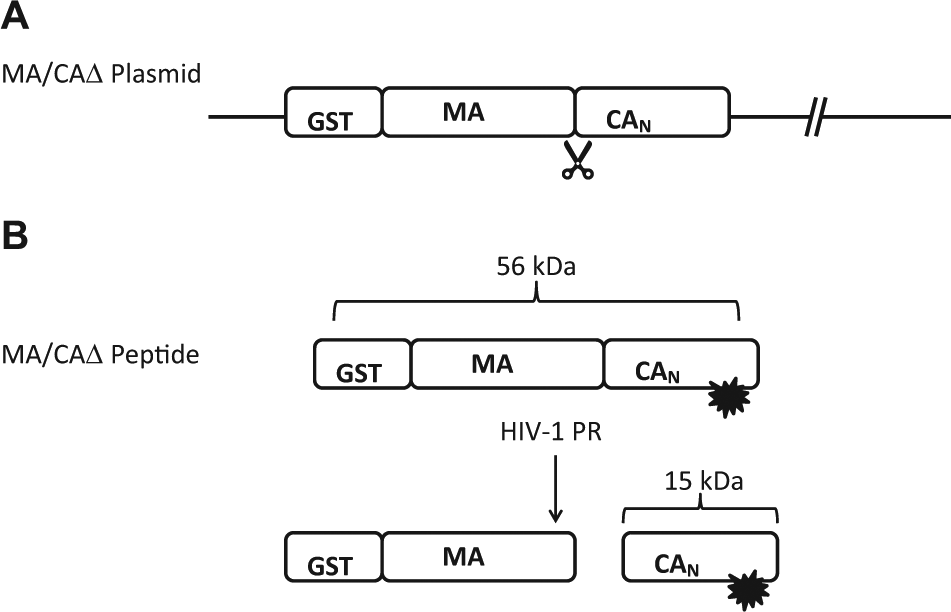
(
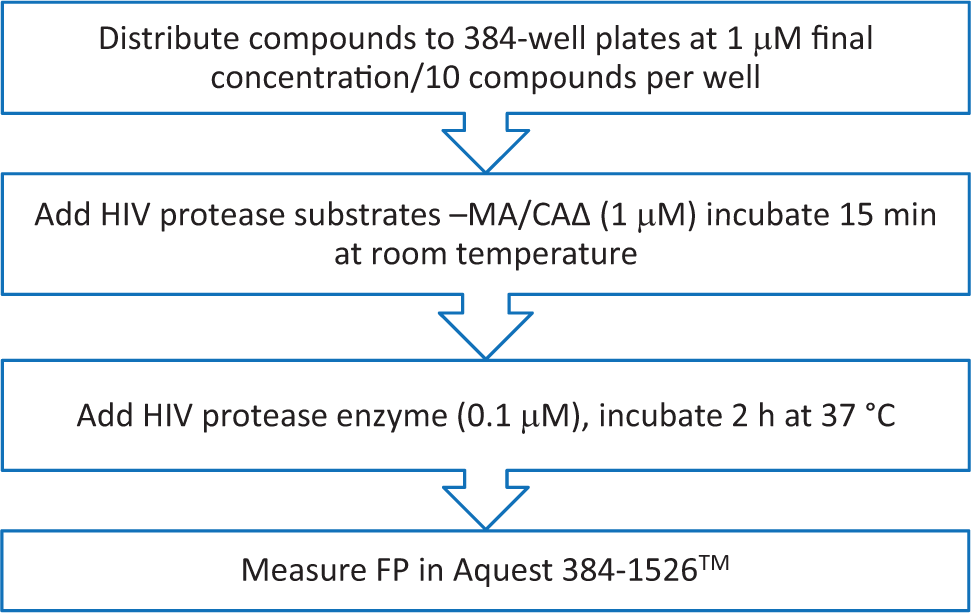
A flowchart of the fluorescent polarization (FP) method employed in the high-throughput screening assay using a 56-kDa MA/CAΔ substrate and human immunodeficiency virus 1 (HIV-1) protease in a 384-well format.
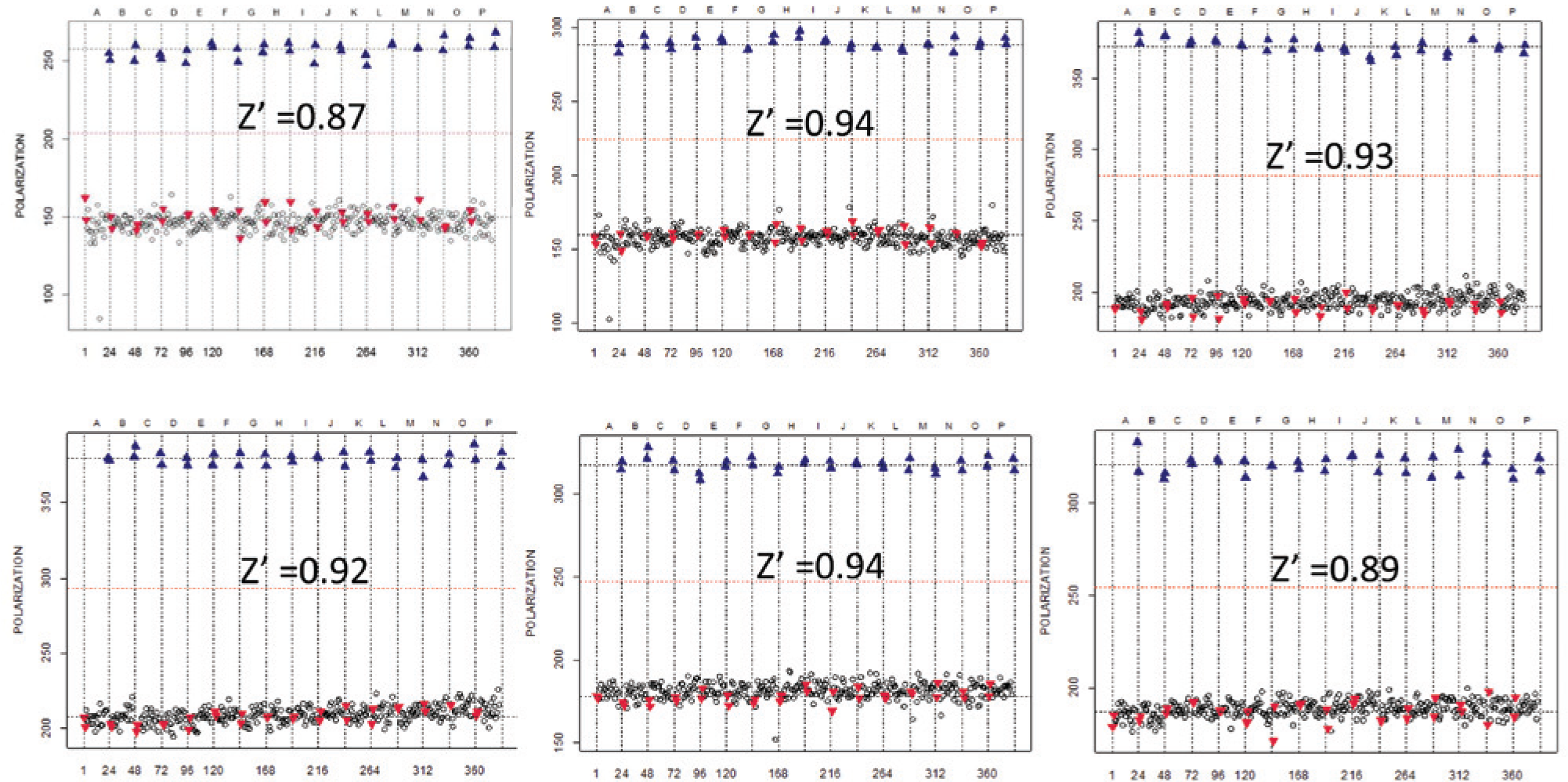
Validation of the high-throughput screening performed over 3 consecutive days. Each day contains a pair of the duplicated assay, each containing 1 µM MA/CAΔ substrate and 100 nM active human immunodeficiency virus 1 (HIV-1) protease in lanes 1 to 2 (high signal control, blue triangles) while lanes 22 to 23 (low signal control, red triangles) contained 100 nM heat-inactivated HIV-1 protease. The Z′ for each assay is shown.
IC50 Determination
We discovered 27 active compounds in the LCGC library after de-convolution of OPL ( Fig. 4 ). Their IC50s were determined using both the MA/CAΔ and a peptide substrate (HIV protease substrate 1) to determine specificity for MA/CAΔ. The same substrate and enzyme concentration as the single-point assay were used for the MA/CAΔ substrate assay, while the inhibitors were serially diluted from 60 µM to 0.003 µM to establish a 10-point curve.

A flowchart of the process that leads to the selection of confirmed compounds to be tested in the secondary gel assay. FP, fluorescence polarization; HIV-1, human immunodeficiency virus 1; HTS, high-throughput screening.
As a counterscreen, we used the HIV protease substrate 1. Unlike the MA/CAΔ substrate, HIV protease substrate 1 is a synthetic peptide with only 12 residues (2% the size of the MA/CAΔ substrate) but retaining the protease cleavage site, Tyr-Pro. In addition, this peptide has two modified amino acids: one is a glutamic acid residue containing the fluorophore EDANS [5-(2-aminoethylamino)-1-napthalene sulfonate], while the other one is a lysine residue modified by the addition of an acceptor chromophore, DABCYL (4′-dimethylamnoazobenzene-4-carboxylate). These two modified residues are on opposite ends of the Tyr-Pro cleavage site. Because of their spatial orientation, DABCYL absorbs the emission of EDAN unless they are separated spatially; after proteolysis at the Tyr-Pro site, such a separation will occur. We believe it is unlikely that those compounds that bind to the complex protein structure of MA/CAΔ would also inhibit HIV protease substrate 1. The resemblance of the 56-kDa MA/CAΔ substrate and the 12 residues of HIV protease 1 are minimal.
Figure 5 shows examples of the IC50 curves for compounds using the MA/CAΔ and HIV protease substrate 1. In all cases, the selected compounds inhibited the enzyme when MA/CAΔ was the substrate, while none were active when HIV protease substrate 1 was used, implying the inhibition was specific to the MA/CAΔ substrate.
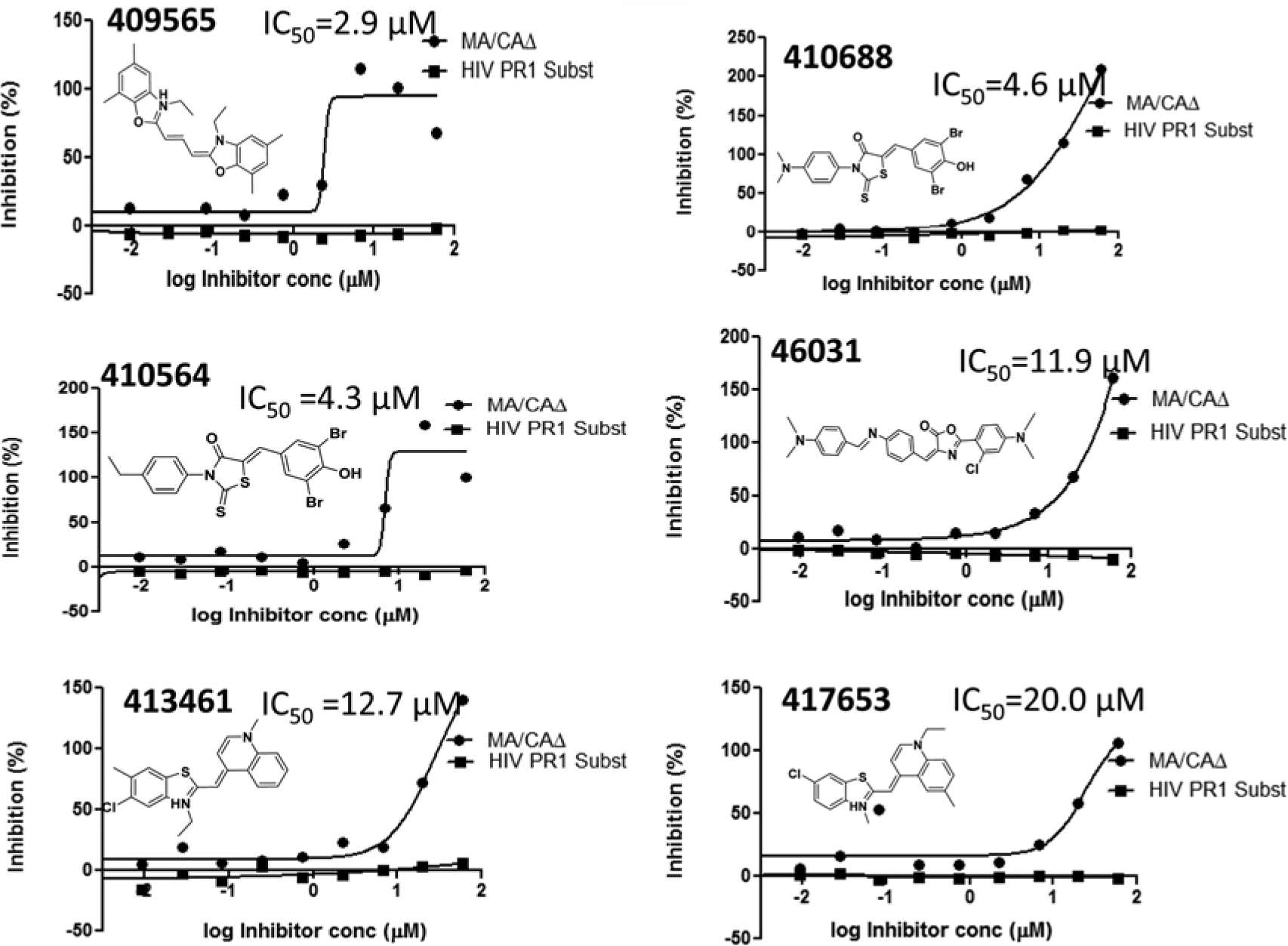
A list of fluorescence polarization assay-confirmed compounds that tested in the secondary gel assay. Their structure and IC50s are shown. HIV, human immunodeficiency virus; PR1, protease substrate 1.
Gel Assay
A gel assay was used to test for biological activity for the most potent compound confirmed as active with the MA/CAΔ substrate. The reaction was identical to the FP assay except the substrate was labeled with 10-fold more FlAsH reagent (2 µM instead of 0.2 µM). Figure 6 shows the gel assays of six different compounds with low IC50s derived from the FP assay. The first six lanes contain samples incubated with the compounds, while lane 7 contains inactivated protease and lane 8 has no inhibitor. Compounds in lanes 1, 2, 4, and 5 showed little to no effect in this assay. The compounds in lanes 3 and 6 show a decrease in the amount of cleaved substrate but also show a loss of the band representing uncleaved MA/CAΔ substrate.
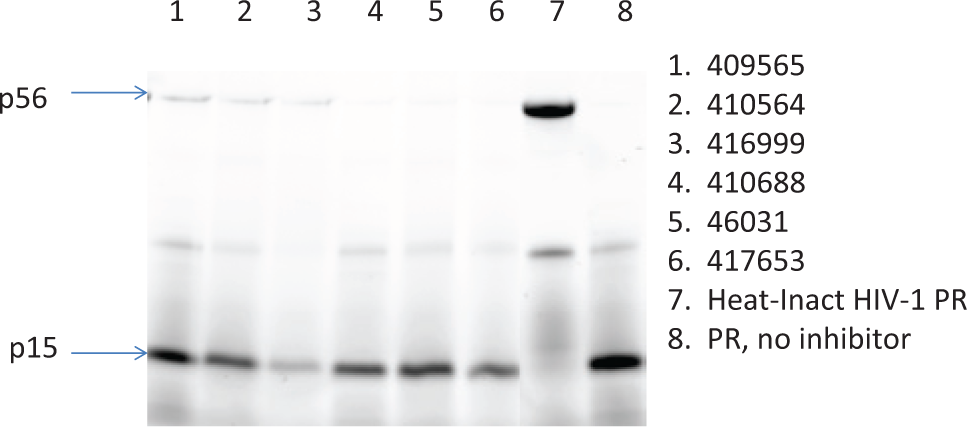
A scan of the gel containing indicated compounds in lanes 1 to 6. The heat-inactivated human immunodeficiency virus 1 (HIV-1) protease (PR) is in lane 7, and the no-compound control is in lane 8. The substrate is the 56-kDa peptide, while the 15 kDa is the substrate.
In conclusion, direct inhibition of the HIV Gag protein interaction represents a novel approach to HIV therapy and a novel target for HTS. The FP assay described here performed well in high throughput with an orthogonally pooled library. While we did find a small number of artifacts in the pooled library, the false-positive rate was sufficiently low to allow the use of this assay in the screening of large libraries. In fact, after de-convolution, the hit rate was only 0.11% (27 actives/24,000 compounds tested). Fifteen of these actives confirmed in potency testing for a 55% confirmation rate.
While FP assays are more resistant to interference from artifacts because of their ratiometric format, compounds that fluoresce at the same wavelength as the detection fluorophore used in the screen have been shown to interfere and cause false-positive results.29,30 This results because the interfering compound is freely rotating and contributes fluorescent signal in the same channels monitored for the screen, thus overwhelming the signal from the fluorescent moiety used in the screen. This produces a signature that is consistent with a decrease in FP, which would indicate inhibition in our assay. Compounds 1, 2, 4, and 5 were found to be strongly fluorescent ( Fig. 7 ) and did not show activity in the gel assay. Thus, we conclude that they were false positive due to innate fluorescence of the compounds.
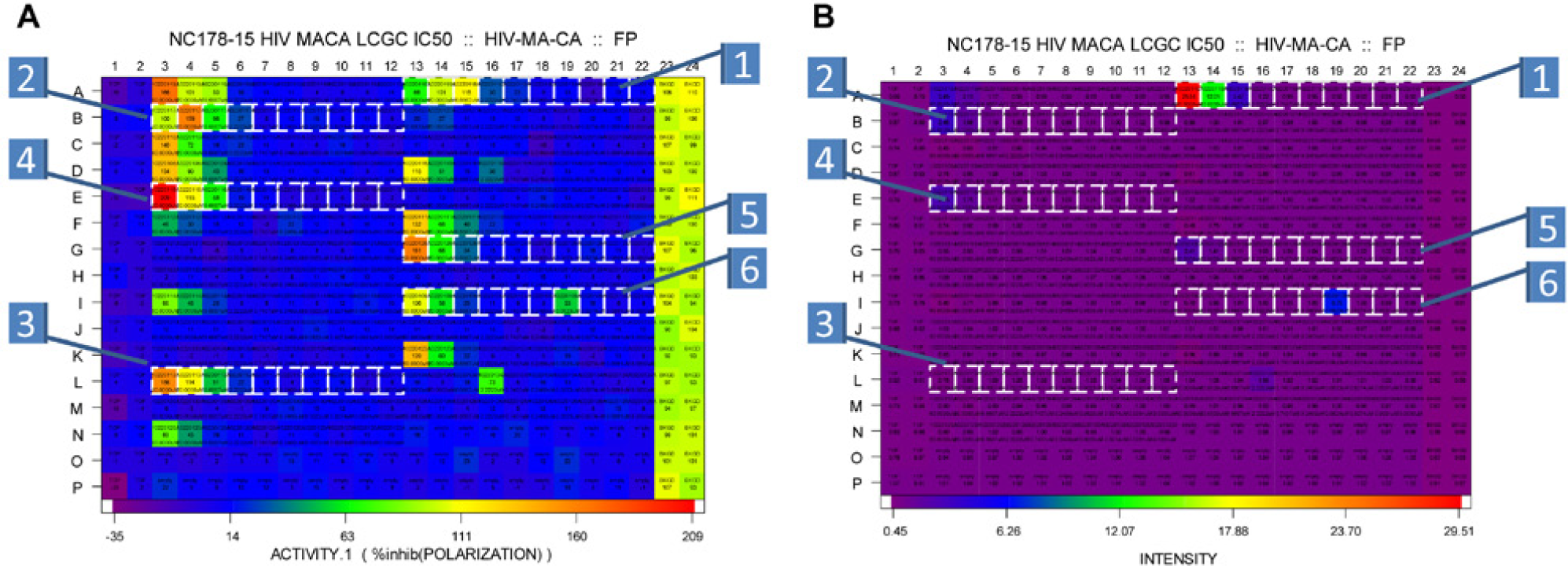
Heat maps of the 384-well assay plates showing the inhibition (
However, fluorescence intensity of the compounds is not the only cause for lack of confirmation from the FP to gel assay. Compounds 3 and 6 decreased the amount of cleaved product produced in the gel assay but did not show an increase in the amount of cleaved substrate. Another well-documented source of false positives in all HTS assays is protein aggregation,30,31 where chemical compounds serve as nucleating agents in the assay. The loss of protein in the gel assay is consistent with this phenomenon.
While none of the compounds discovered in the limited diversity screen here were potent inhibitors of the proteolytic processing of HIV-1 structural protein in the secondary gel assay, the low hit rate in this assay highlights the stringent protein-protein interactions between the HIV-1 protease and the Gag substrate. However, the identification of 15 confirmed hits suggests that physiologically relevant inhibitors can be discovered with this approach. Therefore, we plan to extend our search for inhibitors using new sources of compound diversity.
Footnotes
Acknowledgements
We thank Chatura Jayakody for the preparation of compounds into the 384-well format and Lloyd Frick for helping with the preparation of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by National Institutes of Health (NIH) Grant R21 NS073052.