
Abstract
Study Design
Basic Research.
Objective
Intervertebral disc degeneration (IVDD) is caused by the cartilage endplate (CEP). Cartilage endplate stem cell (CESC) is involved in the recovery of CEP degeneration. Tension load (TL) contributes a lot to the initiation and progression of IVDD. This study aims to investigate the regulatory mechanism of the Mitogen-activated protein kinases/Mammalian target of rapamycin (MAPK/mTOR) pathway during TL-induced CESC degeneration.
Methods
CESCs were isolated from New Zealand big-eared white female rabbits (6 months old). FX-4000T cell stress loading system was applied to establish a TL-induced degeneration model of CESCs. Western blotting was used to detect the level of mTOR pathway-related proteins and autophagy markers LC3-Ⅱ, Beclin-1, and p62 in degenerative CESCs. The expression of MAPK pathway-related proteins JNK and extracellular signal-regulated kinases (ERK) in degenerated CESCs was inhibited by cell transfection to explore whether JNK and ERK play a regulatory role in TL-induced autophagy in CESCs.
Results
In the CESC degeneration model, the mTOR pathway was activated. After inhibition of mTOR, the autophagy level of CESCs was increased, and the degeneration of CESCs was alleviated. The MAPK pathway was also activated in the CESC degeneration model. Inhibition of JNK expression may alleviate TL-induced CEP degeneration by inhibiting Raptor phosphorylation and activating autophagy. Inhibition of ERK expression may alleviate TL-induced CEP degeneration by inhibiting mTOR phosphorylation and activating autophagy.
Conclusion
Inhibition of JNK and ERK in the MAPK signaling family alleviated TL-induced CESC degeneration by inhibiting the phosphorylation of Raptor and mTOR in the mTOR pathway.
Keywords
Introduction
Intervertebral disc degeneration (IVDD) is a common disease closely associated with accelerated or advanced signs of aging, leading to many spinal-related diseases. 1 Cartilage endplate (CEP) is an indispensable structure between the vertebral body and intervertebral disc (IVD), which is conducive to load balance between the vertebral body and intervertebral body. 2 The mechanism of IVDD includes a reduction in the nutrient supply from CEPs to the inner layer of annulus fibrosus and nucleus pulposus cells (NPCs) and impaired CEP-mediated regulation of IVDD-associated anabolism and catabolism, which result in senescence and apoptosis of NPCs. 3 Therefore, CEP degeneration may be a key initiating factor in the pathogenesis of IVDD. 4 Previous studies have illustrated that in human CEP, cartilage endplate stem cells (CESCs) can differentiate into osteoblasts, adipocytes, and chondrocytes, which are very vital for maintaining the structural and functional integrity of CEP.5-7 Moreover, studies have shown that IVDD is not only caused by cell aging but also mainly because of the aging of adult stem cells that fail to timely replenish the aging and damaged cells in the tissues, suggesting that the CESCs in IVD may play an important role in IVDD.8,9 Tension load (TL) contributes a lot to the initiation and progression of IVDD.10,11 Therefore, the in-depth study of the specific mechanism of TL-induced CESCs degeneration is of great significance for the early prevention and treatment of IVDD.
Mitogen-activated protein kinases (MAPKs) are a family of signal transduction molecules involving extracellular signal-regulated kinases (ERKs), stress-activated protein kinase/c-Jun NH(2)-terminal protein kinases, and p38 MAPK, 12 which play a key role in inflammatory responses during IVDD. 13 As a stress sensor, MAPKs pathway is specifically activated in various tissues due to the influence of mechanical stress.14,15 In addition, the MAPK pathway is involved in the occurrence and development of IVD and is essential in the normal differentiation of annulus fibrosus cells.16,17 The phosphorylation of p38 MAPK was increased in CEP under periodic stretch, and p38 MAPK phosphorylation reduced the degeneration and calcification of endplate chondrocytes. 18 And 3% cyclic stretch can participate in the regulation of CEP degeneration by affecting the ERK1/2 pathway. 19 However, the regulatory role and molecular mechanism of MAPK in TL-induced CESC degeneration remain unclear.
Mammalian target of rapamycin (mTOR) is a relatively evolutionarily conserved serine-threonine protein kinase, which is involved in gene transcription and protein expression through phosphorylation of its downstream target protein, thereby affecting autophagy and apoptosis. 20 A study has shown that inhibition of mTOR can strongly induce autophagy. 21 It has been reported that increased autophagy activity in endplate chondrocytes can prevent the apoptosis of endplate chondrocytes, suggesting that mTOR-mediated autophagy can protect the apoptosis of endplate chondrocytes, 22 and inhibition of mTOR can effectively protect articular cartilage tissues. 23 At present, many relevant reports have explored the interaction between MAPK and mTOR pathway, proving that the MAPK pathway may have a regulatory effect on the mTOR pathway.24,25 However, studies on the changes and interactions of the MAPK and mTOR pathways during TL-induced CEP degeneration have not been reported. This study was to explore the mechanism of the MAPK/mTOR pathway during the TL-induced CESC degeneration, identify the key signaling molecules, and further reveal the pathological mechanism of IVDD, so as to provide new ideas and new approaches for the mechanism research and prevention of IVDD.
Materials and methods
Isolation, Extraction, and Culture of Rabbit CESCs
Two specific pathogen-free female white big-eared rabbits (6 months old) from New Zealand were euthanized by intravenous injection of pentobarbital sodium (200 mg/kg) at the ear margin. 26 The fur on the back was removed followed by disinfection with 75% alcohol. The rabbit thoracolumbar spine was removed through the back approach and immediately transferred to a sterile ultra-clean workbench. The T10-L5 IVD segment was collected, and then the surrounding attached muscles, ligaments, and bone fragments were carefully removed and rinsed with phosphate buffer saline (PBS) buffer containing 1000 U/mL penicillin and 1 g/L streptomycin (Gibco, NY, USA). Under the anatomical microscope (SZX16, Olympus, Japan), the annulus fibrosus was incised with a blade, and the nucleus pulposus and annulus fibrosus were scraped away. The CEP with thin middle, thick edge, and transparent shape was put into the petri dish. After washing with the above PBS solution, the CEP was cut into 1 mm3 pieces, detached with 0.25% ethylenediaminetetraacetic acid (EDTA)-trypsin solution (Gibco) and 0.2% type II collagenase (Gibco), and then filtered through a sieve, and endplate chondrocytes were obtained by centrifugation at 600 g for 8 minutes. Cell suspension was prepared with Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (Gibco) containing 20% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and cultured at 37˚C with 5% CO2 to obtain primary endplate chondrocytes.
Stem cells were screened by the principle that low-melting point agarose stem cells can form cell clusters in the suspension culture system.27,28 Agarose (Bio-Rad, Hercules, CA, USA) was dissolved in a PBS solution to obtain agarose at concentrations of 20 g/L and 40 g/L. Agarose (20 g/L) was placed on the bottom layer of the petri dish and solidified at 4°C for 1 hour. Cell suspension (5 × 104 cells/mL) and an equal volume of agarose (40 g/L) were mixed into the petri dish (10% FBS), and condensed at 4˚C for 15 minutes, followed by culture at 37˚C with 5% CO2, and the medium was changed every 2-3 days. Cell clusters formed after 6 weeks and were selected by cell cloning screening system ClonePix2 (Genetix, Molecular Devices, UK) and transferred to 12-well plates and cultured in DMEM/F12 medium (20% FBS) at 37°C with 5% CO2 for amplification in vitro. The obtained cells were subjected to subsequent experimentation.
Flow Cytometry
The well-growing CESCs with a cell density of 80% were prepared into 2 × 106/mL cell suspensions. Cell suspension (500 μL) was incubated with 5 μL monoclonal antibodies FITC-fluorescein-conjugated anti-CD90 (anti-CD90-FITC, ab124527, Abcam), anti-CD105 (anti-CD105-FITC, ab18278, Abcam), and anti-CD34 (anti-CD34-FITC, ab131589, Abcam) in the dark for 20 minutes, with mouse homotype antibodies as control. After the cells were washed in PBS and re-suspended, the cells were detected by flow cytometry immediately.
Identification of Multidirectional Differentiation Ability of CESCs
The CESCs obtained by monoclonal methods were induced to differentiate into adipogenic, osteogenic, and chondrogenic types, respectively. The steps of adipogenic induction and differentiation were as follows 29 : the cells were detached with 0.25% EDTA-trypsin and seeded into 24-well plates at 2 × 104 cells/well, and the adipogenic differentiation medium (DMEM/F12: 10% FBS, 500 μmol/L methylisobutylxanthine, 1 μmol/L dexamethasone, 60 μmol/L indomethacin, and 5 mg/L insulin) was added to induce adipogenic differentiation for 3 days when the cell confluence reached 80-90%. Then the medium was replaced with the maintenance adipogenic differentiation medium (DMEM/F12: 10% FBS, 10 mg/L insulin) for induction of 1 day, and then replaced with adipogenic differentiation medium for induction of 3 days. After 3 cycles, the CESCs were fixed with 4% paraformaldehyde (Hushi, Sinopharm, Shanghai, China), stained with oil red O (Solarbio, Shanghai, China), and observed under a microscope (TS100, Nikon, Japan) and photographed.
The steps of osteogenic induction and differentiation were as follows 30 : after detachment with 0.25% EDTA-trypsin, the cells were seeded into 24-well plates at 2 × 104 cells per well. After 24 h of cell adherence, the medium was replaced with the osteogenic induction differentiation medium (DMEM/F12: 10% FBS, 100 nmol/L dexamethasone, 10 mmol/L β-glycerol phosphate, and 50 μmol/L L-ascorbate-2-phosphate), and changed every 3 days. After 2-3 weeks of continuous induction, the CESCs were fixed with 4% paraformaldehyde, stained with 1% alizarin red (Solarbio), and observed under a microscope and photographed.
The steps of chondrogenic induction and differentiation were as follows 31 : after detachment with 0.25% EDTA-trypsin, the cells were seeded into 24-well plates at 2 × 104 cells/well. The cartilage induction differentiation medium (DMEM/F12: 10% FBS, 1% ITS, 100 nmol/L dexamethasone, 50 mg/L L-ascorbate-2-phosphate, and 10 μg/L recombinant human TGFβ1) was added when the cell confluence reached 80% and changed every 3 days. After 3 weeks of induction, cells were fixed with 4% paraformaldehyde and stained with 1% toluidine blue (Solarbio).
Establishment of CESCs Degeneration Models Induced by TL
The identified rabbit CESCs were subcultured. The cells at the second generation were seeded into BioFlexTM 6-well plate (Flexcell International Corporation, Hillsborough, NC, USA) coated with type I collagen on the surface at 1 × 105 cells/well for culture at 37°C with 5% CO2. When reaching confluence 80-90%, the cells were pretreated with serum-free DMEM for 24 hours. FX-4000T cell stress loading system (Flexcell International Corporation) was used to apply periodic tension strain at 10% elongation and .5 Hz to CESCs for 4 hours/day. The internal environment of the stress loading system was a cell incubator containing 5% CO2 at 37°C. The cells in the control group were seeded in BioFlexTM 6-well plate with the same cell density and continued to be placed in the cell incubator for normal culture without receiving stretching. After 25 days of culture, cells were collected by trypsin digestion for subsequent experimentation.
Cell Treatment and Grouping
The CESCs were cultured in DMEM/F12 medium containing 20% FBS and 5% CO2 at 37°C. The CESCs were divided into (1) control group: CESCs without receiving stretching (NC) (as a control for cells subjected to stretch tension); (2) Tension load group (TL): 0.5 hz and 10% of stretch tension was applied to cells for continuous treatment of 25 days at 4 h/d during cultivation; (3) TL + PBS group: the equal amount of PBS was added to the medium (as a control for cells added with rapamycin), 0.5 hz and 10% of stretch tension were applied for 25 days at 4 h/d; (4) TL + rapamycin group (TL + Rapa): 100 nM rapamycin was added to the medium (553211, Sigma-Aldrich, USA), 32 0.5 hz and 10% of stretch tension were applied for 25 days at 4 h/d; (5) TL + silencing (si)RNA-NC group (TL + si-NC): after transfection of control siRNA (as a control for cells transfected with si-JNK and si-ERK), the cells were subjected to a stretch force of 0.5 hz and 10% for 25 days at 4 h/d; (6) TL + JNK-siRNA group (TL + si-JNK): after transfection with JNK-siRNA, the cells were subjected to a stretch force of 10% and 0.5 Hz for 25 days at 4 h/d; and (7) TL + ERK-siRNA group (TL + si-ERK): after the cells were transfected with ERK-siRNA, a stretch force of 0.5 Hz and 10% was applied for 25 days at 4 h/d days.
Cell Transfection
Well-grown CESCs with a cell density of 80% were transfected with JNK-siRNA (#6232, Cell Signaling Technology, Boston, USA), ERK-siRNA (#6560, Cell Signaling Technology), and negative control SignalSilence® Control siRNA (#6568, Cell Signaling Technology) according to the instructions of Lipofectamine™2000 transfection kit (Gibco). The culture medium was replaced 72 hours later, and the cells were collected for subsequent experimentation. Cell transfection efficiency was detected by reverse transcription quantitative polymerase chain reaction (RT-qPCR).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide Assay
According to the instructions of MTT assay kits (Sigma-Aldrich, MO, USA), cell suspension was prepared from cells in the logarithmic growth stage and laid in the middle well of 96-well plates. The edge wells were filled with sterile PBS and the cells were cultured at 37°C with 5% CO2 for 24 hours. MTT solution (5 mg/mL) was added to each well and cultured at 37°C for 4 hours. Then cells were added with 150 μL dimethyl sulfoxide (DMSO) (Sigma-Aldrich) and mixed evenly to make the crystals fully dissolved. Optical density (OD) value was measured at 490 nm using a spectrophotometer (Bio-Rad 680, Bio-Rad, Hercules, CA, USA). Cell survival rate = [experimental group OD - blank group OD]/(control group OD - blank group OD) × 100%.
Annexin V-FITC/PI Double Staining
CESCs in the logarithmic growth stage were detached with 0.25% trypsin and re-suspended with PBS solution according to the instructions of Annexin V-FITC/propidium iodide (PI) apoptosis kit (YEASEN Biotech, Shanghai, China). Annexin V-FITC (5 μL) and PI (10 μL) staining solutions were added into 100 μL 1 × Binding Buffer, mixed gently, and reacted at room temperature to avoid light for 15 minutes. Flow cytometry (MoFloAstrios EQ, Beckman Coulter, Inc, CA, USA) was used for observation and detection within 1 hour.
Western Blot
Radio immunoprecipitation assay lysis buffer (Beyotime, Shanghai, China) containing protease inhibitor cocktail (Sigma-Aldrich) was added into the cells and mixed. Cells were lysed on ice for 30 minutes, centrifuged at 1600 × g for 10 minutes, and the protein concentration was measured using bicinchoninic acid protein quantitative kits (Boster, Wuhan, China). Then the protein was loaded for boiling, isolated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electrically transferred to polyvinylidene difluoride membranes, which were sealed with 5% bovine serum albumin for 2 hours at room temperature to block the non-specific binding. Next, the membranes were incubated with anti-mTOR (ab134903, 1:1000, Abcam), anti-p-mTOR (Ser2448) (#5536, Cell Signaling Technology), anti-Raptor (ab40768, 1:1000, Abcam), anti-p-Raptor(Ser792) (#8914, Cell Signaling Technology), anti-ERK (ab32537, 1:1000, Abcam), anti-p-ERK (Thr202/Tyr204) (#9101, Cell Signaling Technology), anti-JNK (ab110724, 1:1000, Abcam), anti-p-JNK (Thr183/Tyr185) (#4668, Cell Signaling Technology), anti-p38 (ab170099, 1:1000, Abcam), anti-p-p38 (Thr180/Tyr182) (#4511, Cell Signaling Technology), anti-LC3 II/I (ab128025, Abcam), anti-Beclin-1 (ab207612, Abcam), and anti-p62 (ab109012, Abcam) at 4°C overnight. After washing the membrane, goat anti-rabbit IgG labeled with horseradish peroxidase (ab205718, 1:2000, Abcam) was added for incubation for 2 hours. Finally, the developing images were collected by ImageJ software (NIH, Bethesda, MD, USA), with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal reference.
RT-qPCR
Primer Sequence of RT-qPCR.

Statistical Analysis
SPSS 21 (IBM Corp. Armonk, NY, USA) and GraphPad Prism 8.01 (GraphPad Software Inc., San Diego, CA, USA) were used for statistical analysis and plotting. The t test was used for data comparison between 2 groups. One-way analysis of variance (ANOVA) was used for data comparison among multiple groups, and Tukey’s test was used for the post hoc test. A probability value of P < .05 indicated the difference was statistically significant.
Results
CEP Degeneration Induced by TL
First, the rabbit CESCs were isolated and extracted. Flow cytometry showed positive expression of stem cell markers CD90 and CD105 and negative expression of CD34 (Figure 1A), indicating that CESCs were successfully obtained from rabbit CEP. To further verify whether rabbit CESCs have the biological differentiation ability of stem cells, we conducted the three-way differentiation induction of adipogenesis, osteogenesis, and chondrogenesis. The results manifested that rabbit CESCs have the differentiation ability of adipogenesis, osteogenesis, and chondrogenesis (Figure 1B). These results indicate that we have successfully obtained the rabbit CESCs. To deliberate the effect of TL on CESCs, we applied the FX-4000T cell stress loading system to impose 10% elongation and 0.5 Hz periodic tension strain to cells. Cell viability was determined by MTT assay, which elicited decreased cell viability of the TL group compared with the NC group (P < .01) (Figure 1C). Annexin V-FITC/PI double staining showed higher apoptosis levels in the TL group than the NC group (P < .01) (Figure 1D). The expressions of cartilage markers proteoglycan ACAN, type II collagen COL-2A, and transcriptional regulator Sox9 were detected by RT-qPCR, which elucidated decreased ACAN, COL-2A, and SOX9 expressions in the TL group compared to the control group (all P < .01) (Figure 1E), indicating the degeneration of CESCs induced by TL. TL-induced degeneration of Cartilage endplate stem cells. (A): The expression of CD90, CD105, and CD34 were detected by flow cytometry; (B): rabbit cartilage endplate stem cells were stained after osteogenic, adipogenic, and chondrogenic differentiation induction; (C): cell activity was detected by MTT assay; (D): apoptosis was detected by Annexin V-FITC/PI double staining assay; E: mRNA expressions of cartilage markers ACAN, COL-2A, and Sox9 were detected by RT-qPCR. Cell experiment was conducted 3 times. Data was expressed as mean ± standard deviation. The t test was used for data comparison between the 2 groups. ** P < 0.01, *** P < 0.001.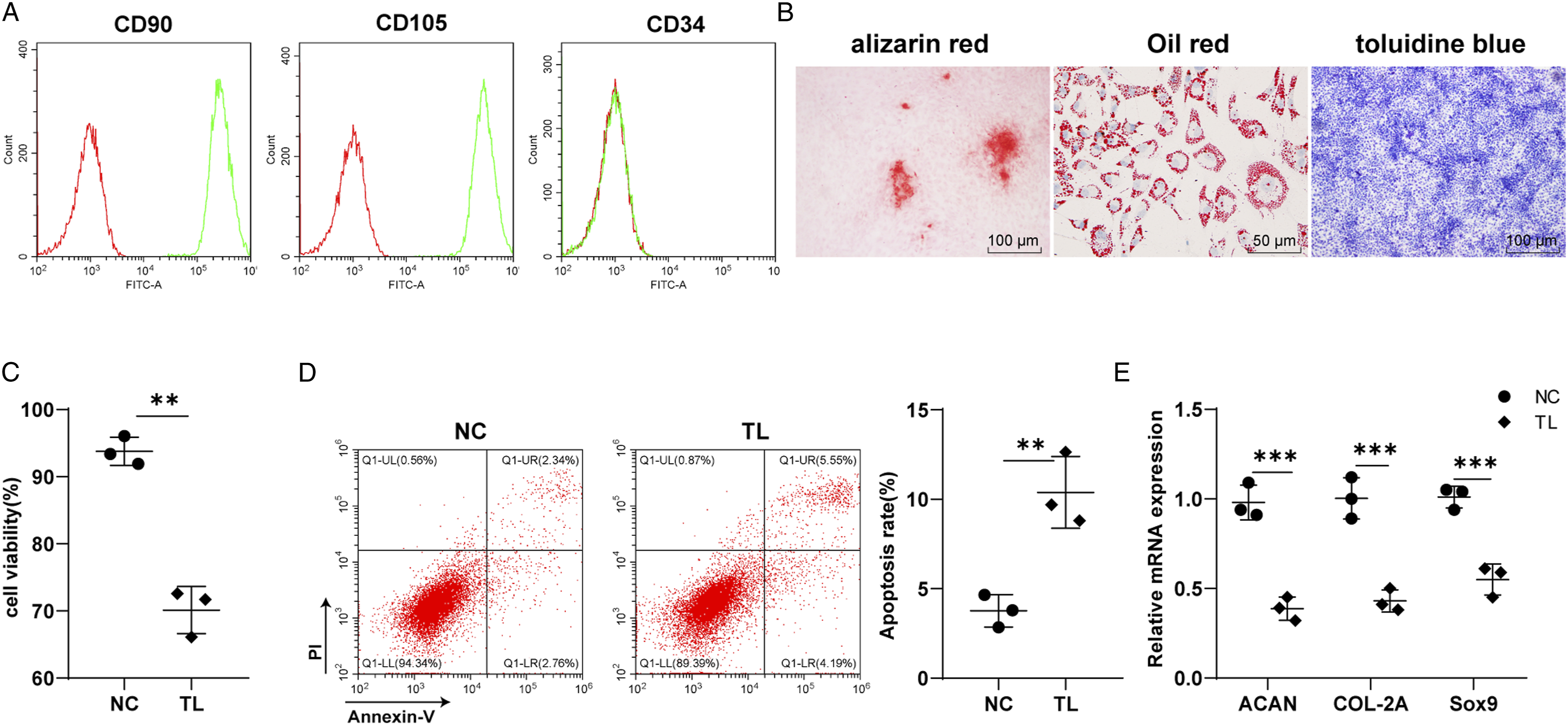
Inhibition of the mTOR Pathway Alleviated TL-Induced CEP Degeneration by Activating Autophagy
To ascertain whether the mTOR pathway is involved in the degeneration of CESC under TL, we detected the protein expressions of mTOR, p-mTOR, Raptor, and p-Raptor in the mTOR pathway by Western Blot (WB). The results elicited that the ratio of p-mTOR/mTOR and the ratio of p-Raptor/Raptor were significantly increased (all P < .01) (Figure 2A), indicating the activation of phosphorylation in the mTOR pathway. Inhibition of the Mammalian target of rapamycin pathway alleviates TL-induced cartilage endplate degeneration by activating autophagy. (A): protein levels of Mammalian target of rapamycin, p-mTOR, Raptor, and p-Raptor after adding rapamycin were detected by Western Blot; (B): cell activity was detected by MTT assay; (C): apoptosis was detected by Annexin V-FITC/PI double staining assay; (D): mRNA expressions of cartilage markers ACAN, COL-2A, and Sox9 were detected by RT-qPCR; (E): Western Blot was used to detect the protein levels of autophagy markers LC3-II, Beclin-1, and p62 after rapamycin addition. Cell experiment was conducted 3 times. Data was expressed as mean ± standard deviation. One-way ANOVA was used for data comparison among multiple groups and Tukey’s test was used for the post hoc test. * P < .05, ** P < .01, *** P < .001.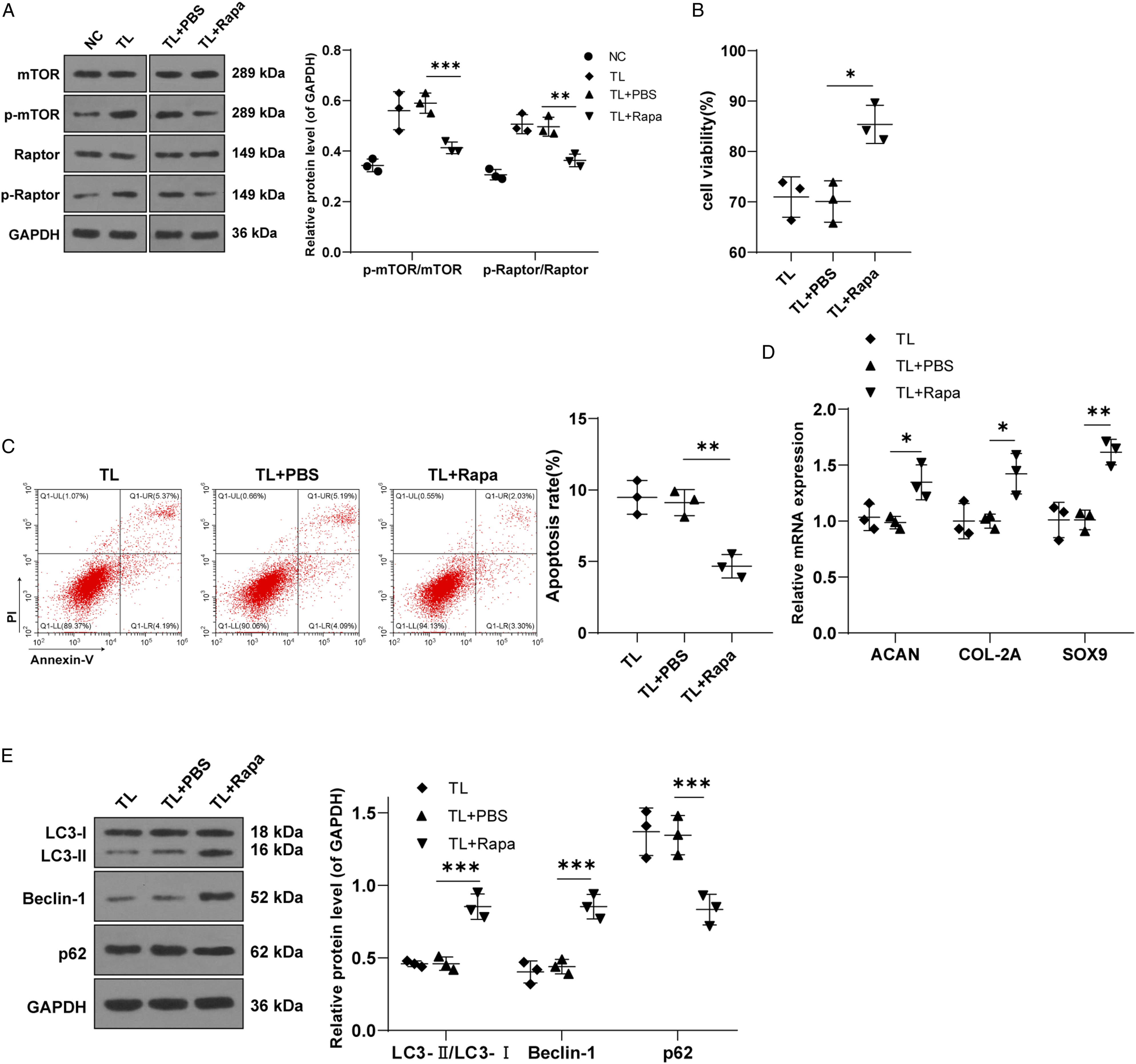
Rapamycin is frequently used as a specific inhibitor of the mTOR pathway. 32 Therefore, we further verified the influence of the mTOR pathway on the degeneration level of CESCs after TL by adding 100 nM rapamycin to the medium. Western blot showed that the ratios of p-mTOR/mTOR and p-Raptor/Raptor were lowered after the addition of rapamycin (all P < .05), indicating the suppression of the mTOR pathway (Figure 2A). Furthermore, MTT assay was used to detect cell viability, which discovered that compared with the TL + PBS group, cell viability was increased in the TL + Rapa group (P < .01) (Figure 2B). Annexin V-FITC/PI double staining showed that the apoptosis level of the TL + Rapa group was lower than the TL + PBS group (P < .01) (Figure 2C). Subsequent RT-qPCR showed that the levels of cartilage markers ACAN, COL-2A, and SOX9 in CESCs were up-regulated after adding rapamycin (all P < .05) (Figure 2D). These results suggest that inhibition of the mTOR pathway can alleviate TL-induced CEP degeneration. To figure out whether the mTOR pathway regulates CESCs degeneration through autophagy, we detected the protein levels of autophagy markers LC3-II, Beclin-1, and p62 by WB. The results demonstrated that the levels of LC3-II and Beclin-1 in the TL-Rapa group were up-regulated, and p62 was reduced (all P < .05) (Figure 2E), suggesting that the addition of rapamycin could activate autophagy of CESCs. Taken together, down-regulation of the mTOR pathway may alleviate TL-induced CEP degeneration by activating autophagy.
The MAPK Pathway Was Stimulated in CESCs After TL
Presently, cumulative studies indicate that the MAPK pathway may regulate the mTOR pathway.24,25 First, the key proteins ERK, JNK, p38, p-ERK, p-JNK, and p-p38 of the MAPK pathway in CESCs under TL were detected by WB. Interestingly, compared with the NC group, the protein expressions of p-ERK, p-JNK, and p-p38 in the TL group were visibly enhanced (all P < .05), while there were no significant differences in protein expressions of ERK, JNK, and p38 (all P > .05) (Figure 3), indicating that the MAPK pathway was activated in CESCs after TL. Activation of the Mitogen-activated protein kinases pathway in the model of CESCs degeneration induced by TL. The protein levels of ERK, p-ERK, JNK, p-JNK, p38, and p-p38 were detected by Western Blot. Cell experiment was conducted 3 times. Data was expressed as mean ± standard deviation. The t test was used for data comparison between the 2 groups. ** P < 0.01, *** P < 0.001.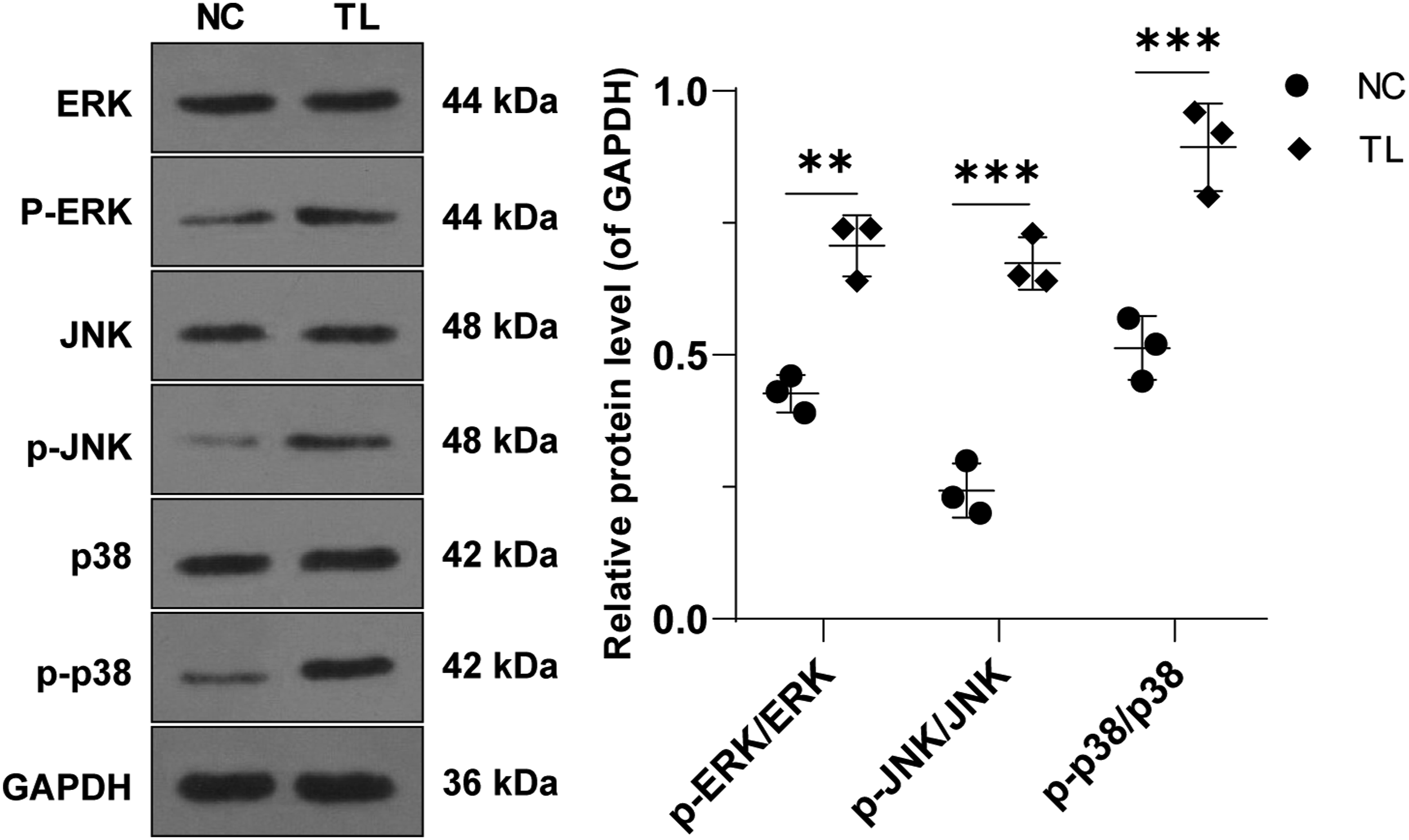
JNK Knockdown Alleviated CESC Degeneration by Inhibiting Phosphorylation of Raptor
JNK, a key protein of the MAPK pathway, phosphorylates Raptor, a key regulatory part of mTORC1.
33
The molecular association between JNK and Raptor may be a potential regulatory mechanism between the MAPK and mTOR pathways.34,35 Therefore, we further studied the regulation of JNK and Raptor in TL-induced CESC degeneration. To begin with, si-JNK and its negative control si-JNK-NC (si-NC) were transfected into TL-treated CESCs, and the mRNA expression of JNK was detected by RT-qPCR. The results discovered that the JNK expression in TL-treated CESCs was successfully repressed after transfection with si-JNK (P < .01) (Figure 4A). Western blot showed that the p-Raptor/Raptor ratio in the TL + si-JNK group was decreased (P < .05) (Figure 4B), indicating that the JNK pathway regulates Raptor phosphorylation. To further investigate the role of JNK in regulating Raptor phosphorylation in CESCs degeneration under TL, we performed MTT assay and Annexin V-FITC/PI double staining. The results uncovered that compared with the TL + si-NC group, si-JNK transfected cells showed enhanced cell activity and decreased apoptosis level (P < .01) (Figures 4C and 4D). Subsequent RT-qPCR showed that the levels of cartilage markers ACAN, COL-2A, and SOX9 were up-regulated in the TL + si-JNK group compared to the TL + si-NC (all P < .05) (Figure 4E). These results suggest that inhibition of JNK can reduce TL-induced CEP degeneration by down-regulating Raptor phosphorylation. To investigate whether JNK-mediated phosphorylation of Raptor affects the degeneration of CESCs by regulating autophagy, we detected the protein levels of autophagy markers LC3-II, Beclin-1, and p62 by WB. The results revealed that the levels of LC3-II and Beclin-1 in the TL + si-JNK group were up-regulated and p62 was reduced (all P < .05), indicating that inhibition of JNK expression can activate autophagy of TL-treated CESCs (Figure 4F). In conclusion, down-regulation of JNK may activate autophagy by inhibiting Raptor phosphorylation, thereby alleviating TL-induced CEP degeneration. JNK knockdown alleviates CESCs degeneration by inhibiting phosphorylation of Raptor. After transfection of si-JNK/si-NC in TL-induced CESCs, (A): mRNA level of JNK in CESCs after transfection of si-JNK was detected by qRT-PCR; (B): changes in total Raptor and p-Raptor protein levels were detected by WB; (C): cell activity was detected by MTT assay; (D): apoptosis was detected by Annexin V-FITC/PI double staining assay; (E): mRNA expressions of cartilage markers ACAN, COL-2A, and Sox9 were detected by RT-qPCR; (F): WB was used to detect the protein levels of autophagy markers LC3-II, Beclin-1, and p62. Cell experiment was conducted 3 times. Cell experiment was conducted 3 times. Data was expressed as mean ± standard deviation. One-way ANOVA was used for data comparison among multiple groups and Tukey’s test was used for the post hoc test. * P < .05, ** P < .01, *** P < .001.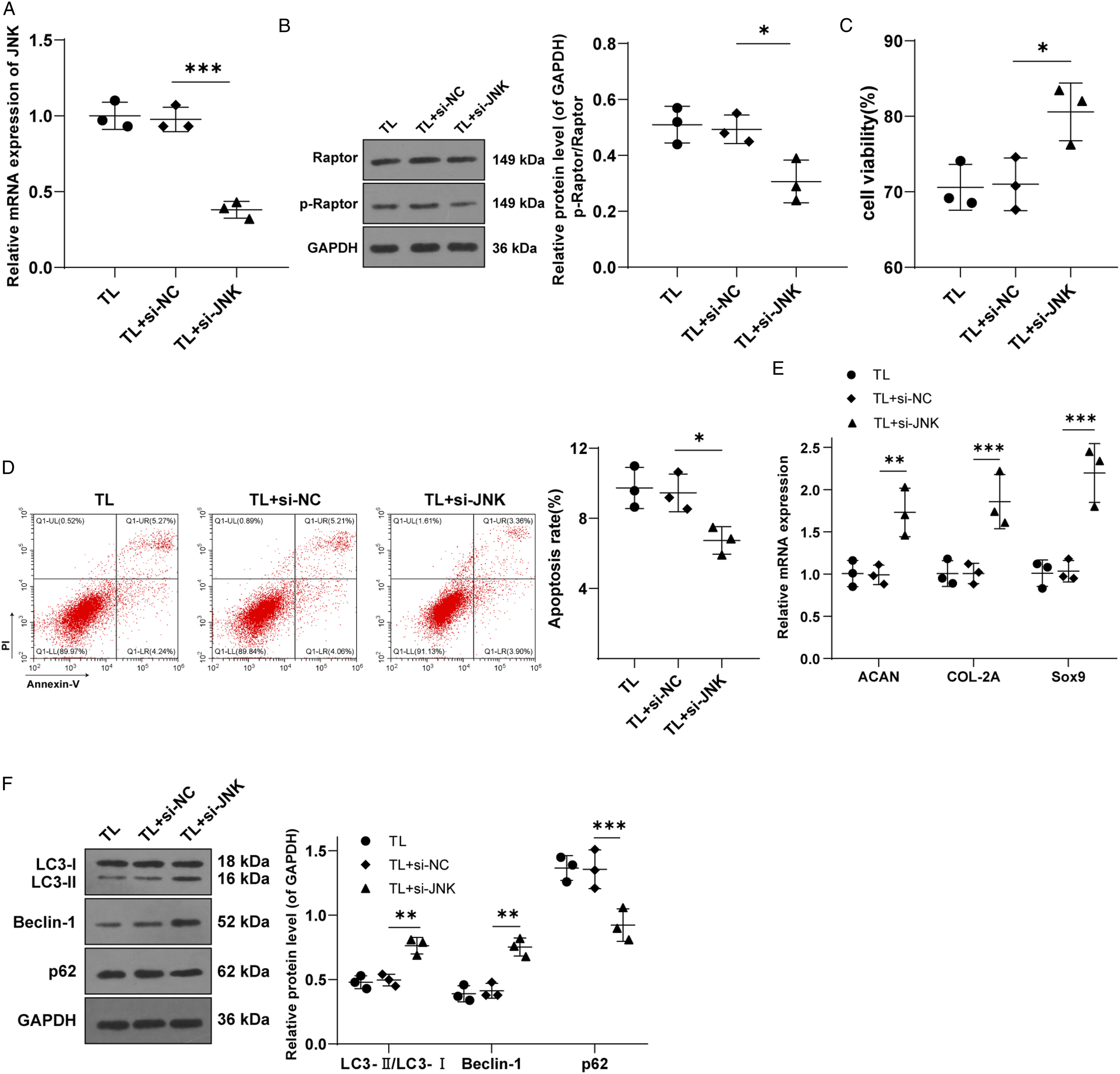
ERK Knockdown Alleviates CESC Degeneration by Inhibiting Phosphorylation of mTOR
In addition to the mechanism of JNK regulating Raptor phosphorylation, there may be many other regulatory mechanisms between the MAPK and mTOR pathways. It has been reported that ERK, a key protein of the MAPK pathway, regulates cell proliferation by mediating the phosphorylation of mTOR.
36
To explore whether ERK regulates mTOR phosphorylation during CESC degeneration, we transfected TL-induced cells with si-ERK or si-ERK-NC (si-NC). The mRNA level of ERK was detected by RT-qPCR, and the results showed that the level of ERK in the TL + si-ERK group was lower than that of the TL + si-NC group (P < .05) (Figure 5A), indicating that ERK in TL-treated CESCs was successfully inhibited. Western blot showed that the ratio of p-mTOR/mTOR was obviously decreased (P < .05) (Figure 5B), indicating that ERK regulates mTOR phosphorylation in CEP degeneration models. To further explore the role of ERK in regulating the mTOR phosphorylation in the degeneration of TL-induced CESCs, we conducted MTT assay and Annexin V-FITC/PI double staining. Compared with the TL + si-NC group, the cell activity was elevated and apoptosis level was decreased in the TL + si-ERK group (Figures 5C and 5D). RT-qPCR showed up-regulated levels of cartilage markers ACAN, COL-2A, and SOX9 in the TL + si-ERK group compared with the TL + si-NC group (all P < .05) (Figure 5E). These results elicited that inhibition of ERK can reduce TL-induced CEP degeneration by down-regulating the phosphorylation of mTOR. We further detected the protein levels of autophagy markers LC3-II, Beclin-1, and p62 by WB. The results manifested that the levels of LC3-II and Beclin-1 in the TL + si-ERK group were up-regulated and p62 was reduced (all P < .05) (Figure 5F), indicating that suppression of ERK expression can activate autophagy of TL-treated CESCs. Overall, inhibition of ERK expression may alleviate TL-induced CEP degeneration by inhibiting the phosphorylation of key proteins in the MAPK pathway and activating autophagy. ERK knockdown alleviates CESCs degeneration by inhibiting phosphorylation of mTOR. After transfection of si-JNK/si-NC in TL-induced CESCs, (A): mRNA level of ERK in CESCs after transfection of si-ERK was detected by RT-qPCR; (B): WB was used to detect the changes in Mammalian target of rapamycin and p-mTOR protein levels after ERK inhibition; (C): cell activity was detected by MTT assay; (D): apoptosis was detected by Annexin V-FITC/PI double staining assay; (E): mRNA expressions of cartilage markers ACAN, COL-2A, and Sox9 were detected by RT-qPCR; (F): WB was used to detect the protein levels of autophagy markers LC3-II, Beclin-1, and p62 after rapamycin addition. Cell experiment was conducted 3 times. Data was expressed as mean ± standard deviation. One-way ANOVA was used for data comparison among multiple groups and Tukey’s test was used for the post hoc test. *** P < .001, ** P < .01, * P < .05.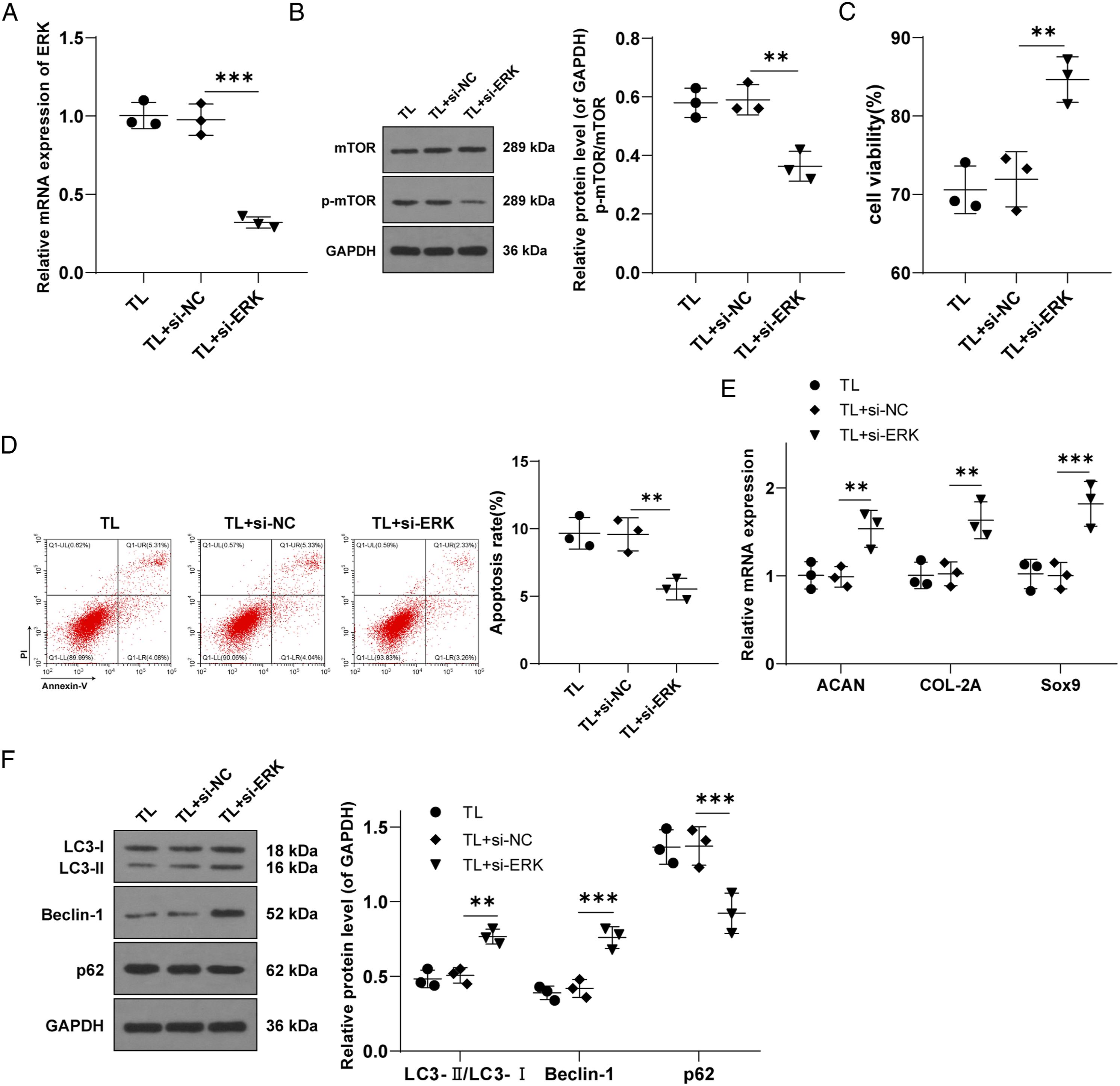
In summary, inhibition of the expression of key proteins JNK and ERK in the MAPK pathway alleviated the TL-induced CEP degeneration by inhibiting the phosphorylation of Raptor and mTOR in the mTOR pathway and activating autophagy.
Discussion
IVDD is considered an imperative cause of low back pain, which affects approximately 80% of adults at various stages of their lives. 37 Mechanical stress is critical to the occurrence and progression of IVDD. 38 Mitogen-activated protein kinases and mTOR have been reported to be associated with endplate chondrocytes and autophagy in IVDD.13,22 Our finding demonstrated that inhibition of JNK and ERK in the MAPK family alleviated TL-induced CESC degeneration by inactivating the phosphorylation of Raptor and mTOR in the mTOR pathway.
This study employed a cell mechanical loading device. The cells adhering to the membrane were imposed with cyclic stretch stimulation through stretching the membrane. We first isolated and extracted rabbit CESCs and established a TL-induced CESC degeneration model. Apoptosis and autophagy are critical in the occurrence and development of IVDD. 39 Autophagy promotes cell adaptation to pressure and may play a central role in regulating IVD cell adaptation to pressure, suggesting that autophagy regulation may be a potential treatment for IVDD. 40 Enhanced autophagy activity can avoid apoptosis of endplate chondrocytes, and the suppression of mTOR can intensely lead to autophagy.41,42 To figure out whether the mTOR pathway is related to the TL-induced degeneration of CESCs, we determined the protein expression of the mTOR pathway. Our results demonstrated that the proportions of p-mTOR and p-Raptor in the total protein of mTOR and Raptor were enhanced after TL treatment. Due to mechanical stimulation, the ratio of upstream to downstream proteins of the mTOR pathway tends to be more phosphorylated than non-phosphorylated. 43 As a whole, the phosphorylation of the mTOR pathway was stimulated. As a specific inhibitor of the mTOR pathway, rapamycin can block the mTOR pathway and regulate autophagy levels. 32 As expected, with the addition of rapamycin, the cell activity was enhanced, and apoptosis levels were reduced, which was consistent with the findings in a previous study that suppression of the mTOR pathway protects IVD cells from apoptosis and degeneration. 44 Additionally, intermittent cyclic mechanical tension induced calcification of endplate chondrocytes and reduced the levels of chondrogenic genes COL-2A, ACAN, and SOX9. 45 Interestingly, our results unraveled that the levels of ACAN, COL-2A, and SOX9 in CESCs were significantly up-regulated after the addition of rapamycin. Inhibition of mTOR can effectively protect articular cartilage and improve pressure-induced IVDD.23,46 Overall, down-regulation of the mTOR pathway alleviated TL-induced CEP degeneration. Next, we observed that the protein levels of autophagy were elevated after the addition of rapamycin. Down-regulation of the mTOR strongly induced autophagy, while increased autophagy activity alleviated IVDD in endplate chondrocytes.21,47 In short, inhibition of the mTOR pathway may alleviate TL-induced CEP degeneration by stimulating autophagy.
The MAPK pathway in endplate chondrocytes was influenced under mechanical force. 15 The phosphorylation levels of MAPKs (ERKs, JNKs, and p38 MAPK) in vascular smooth muscle cells were raised under stretch stress. 48 Consistently, our results revealed the activated MAPK pathway in CESCs after TL treatment. Previous studies have reported that the MAPK pathway can be involved in mechanical stress-induced CEP degeneration by mediating mitochondrial apoptosis.49,50 Meanwhile, the MAPK pathway may regulate the mTOR pathway.24,25The MAPK/JNK pathway is required for both mTOR activation and Raptor phosphorylation. 34 We then silenced JNK in TL-induced CESCs and found that after JNK silencing, the proportion of p-Raptor in the total protein of Raptor was lowered, indicating that the JNK pathway regulates Raptor phosphorylation. Moreover, silencing JNK elevated cell activity, reduced apoptosis, and up-regulated the levels of cartilage markers and autophagy. Prior studies have reported that JNK inhibitors can enhance autophagy of rat NPCs 51 and inhibition of the JNK pathway induced cell activity and reduced apoptosis by enhancing MTF1 nuclear translocation, thus improving IVDD. 52 In brief, inhibition of JNK may alleviate TL-induced CEP degeneration through inhibition of Raptor phosphorylation and activation of autophagy.
ERK controls cell proliferation by mediating mTOR phosphorylation in keratinocytes. 36 High-frequency electrical stimulation reveals ERK-mTOR signaling correlated with force-time integral. 53 To elucidate the regulation mechanism of ERK-mediated mTOR phosphorylation in the regression of CESCs induced by TL, we first silenced ERK by si-ERK transfection. After ERK expression was inhibited, the proportion of p-mTOR in the total protein of mTOR was decreased, suggesting that ERK regulated the phosphorylation of mTOR in the CESC degeneration model. Further, we observed that after silencing ERK, the cell activity was elevated, apoptosis level was decreased, the levels of cartilage markers were up-regulated, and autophagy was activated. ERK inhibition prevented mTOR activation, thus effectively protecting articular cartilage tissue.23,54 Inhibition of ERK prevents early osteoarthritis by activating autophagy. 55 In a word, inhibition of ERK may alleviate TL-induced CEP regression by inhibiting mTOR phosphorylation and activating autophagy.
In conclusion, this study revealed that down-regulation of JNK and ERK-mediated phosphorylation of Raptor and mTOR in the MAPK pathway alleviated TL-induced CESC degeneration. However, the precise regulatory mechanism of the mTOR pathway under TL has not been thoroughly studied. In addition, in this study, the setting of the control group was too simple, and the influence of different tensile forces on CESCs was not explored. On the other hand, the concentration gradient of rapamycin was not set to explore the effects of different doses of rapamycin on alleviating CESC degeneration induced by TL. The specific effects of different TLs and different concentrations of rapamycin on CESC degeneration can be further explored in the future. Another limitation of this study is that in vivo experiments were not conducted to verify the therapeutic effect, which should be supplemented in the future. Meanwhile, we did not further study p38 in the process of exploring key molecules of the MAPK and mTOR pathways. The mechanism of p38 between the 2 pathways will be further explored in the future. Furthermore, according to previous studies, autophagy plays a central role in the adaptation of IVD cells to different extracellular microenvironmental stresses. 40 Starvation, acid, hypoxia, hyperglycemia, and mechanical load can activate autophagy, suggesting that autophagy activation may be a common adaptive mechanism of stressed IVD cells. Further exploration of the MAPK pathway involved in IVD autophagy, especially in vivo studies, is of guiding significance for finding possible useful therapeutic targets for IVD. In the future, we need to further study the precise regulation mechanism of the mTOR pathway under TL to find new targets for the treatment of CEP degeneration and IVDD from the perspective of genes.
Supplemental Material
sj-jpg-1-gsj-10.1177_21925682221085226 – Supplemental Material for Mechanism of the Mitogen-Activated Protein Kinases/Mammalian Target of Rapamycin Pathway in the Process of Cartilage Endplate Stem Cell Degeneration Induced by Tension Load
Supplemental Material, sj-jpg-1-gsj-10.1177_21925682221085226 for Mechanism of the Mitogen-Activated Protein Kinases/Mammalian Target of Rapamycin Pathway in the Process of Cartilage Endplate Stem Cell Degeneration Induced by Tension Load by Yu Zhang, Chen Liu, Yu Li and Hongguang Xu in Global Spine Journal
Footnotes
Authors' contributions
YZ contributes to the definition of learning concepts, learning design, and knowledge content; CL participated in editing and reviewing the manuscript; YL has contributed to experimental research; HGX is responsible for data acquisition and analysis; YZ and CL contributed to statistical analysis. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (grant nos. 82172427, 81972108) and Funding of “Panfeng” Innovation Team Project and “Peak” Training Program for Scientific Research of Yijishan Hospital, Wannan Medical College (grant nos. PF2019007, GF2019T02, GF2019G07, GF2019G12).
Ethics Approval
The experiment was approved by The Yijishan Hospital Academic Ethics Committee. All procedures are carried out in strict accordance with the code of ethics. They made great efforts to reduce the suffering of animals.
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.