
Abstract
Prior to enactment of the final investigational new drug application (IND) safety reporting rule, an attempt was made to document the effort expended at investigative sites in processing IND safety reports from sponsors and to assess the effect of these expedited reports on trial conduct. Investigators were asked to (1) prospectively document time to process IND safety reports and (2) retrospectively review safety reports from a previous 3-month period, documenting resultant actions. In this limited sample, sites spent a median of 0.25 hours per report at a median cost of US$22. Few expedited safety reports were retrospectively said to have changed study conduct or informed consent. However, a low response rate and the concentration of clinical sites in a single therapeutic area preclude generalizing these results. The authors discuss the challenges in gaining investigators’ cooperation to evaluate the impact of regulatory requirements. Better methods to facilitate this type of research will enrich the scientific basis of future clinical trial regulation and guidance.
Keywords
Introduction
Before the US Food and Drug Administration’s (FDA’s) final rule on safety reporting for drugs being studied under an investigational new drug application (IND) went into effect on March 28, 2011, 1 many site investigators participating in multicenter clinical trials conducted under an IND had raised concerns about the number and interpretability of expedited safety reports that they received from study sponsors. For example, over a 6-month interval in 2008, a group of oncology investigators from a single site reported receiving an average of 190 new IND safety reports per month in trials of 2 chemotherapy agents. However, less than 1% of these reports (11 of 1144) met appropriate threshold criteria as unanticipated problems to be forwarded to the institutional review board (IRB) (unpublished data, Bruce Burnett, PhD, Duke Translational Medicine Institute, 2008). Similarly, a group of international investigators noted that although significant effort is expended to process expedited reports of serious adverse reactions, rarely do the reports substantively inform their understanding of a product’s safety profile. 2
However, despite anecdotal accounts of problems with the existing system of expedited safety reporting by sponsors to site investigators, there was little evidence that could be used to objectively characterize the system, study its effects, and serve as a basis for future improvements. Prompted by these concerns, in 2009 the Clinical Trials Transformation Initiative 3 attempted to gather empirical evidence about the system then in effect for sponsors to report serious, unexpected, and associated adverse events to site investigators conducting multicenter IND studies. One component of this effort also sought to explore differences in safety reporting approaches allowed by the US FDA versus the European Commission (EC) and to evaluate whether these different approaches resulted in differential impact on investigators.
Methods
In the present article, we report findings from 2 segments (Objectives 2 and 3) of a larger project examining multiple aspects of expedited safety reporting from sponsors to site investigators (http://www.ctti-clinicaltrials.org/what-we-do/study-conduct/adverse-event-reporting).
Prospective Documentation of Resource Requirements
A descriptive sample of 12 to 15 investigators was sought from academic and community sites that were engaged in at least 2 clinical trials being conducted under an IND. A total of 375 investigators were solicited, primarily via e-mail, to participate in the study. Extrapolating from our single-center experience of anecdotal complaints received from clinical investigators about their frustrations with expedited safety reports (unpublished data, Bruce Burnett, PhD, Duke Translational Medicine Institute, 2008), we initially assumed that site investigators would volunteer to participate in our study without compensation. However, when we were unable to achieve adequate participation, we added a gift certificate for US$100 as an incentive for site personnel to document their activities.
To avoid recall bias, we asked investigators to prospectively record the time that they and their staff spent in receiving, interpreting, and communicating expedited safety reports from IND sponsors. Participants were provided with a single-page paper form (Figure 1) to be kept at the workstation of the person (or persons) responsible for processing expedited safety reports. Using this form, investigators were asked to document over a period of 8 weeks the number of individual expedited safety reports received, the time required for managing the reports, and the type of personnel involved in interpreting and communicating relevant information to other staff and to the IRB.
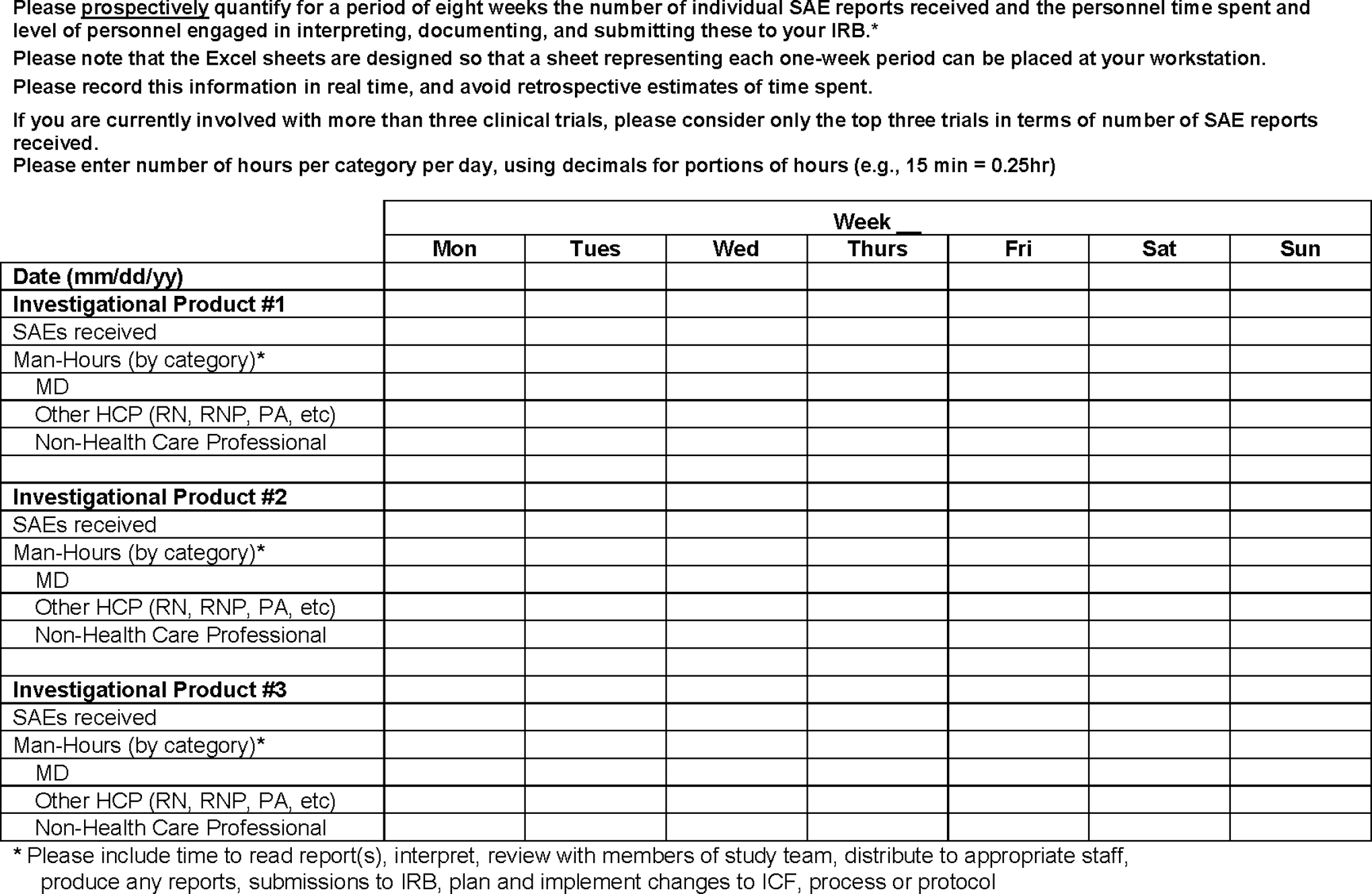
Prospective data form—resource requirements.
Retrospective Documentation of Effect on Trials
In a separate exercise, these same investigators were asked to retrospectively review all expedited safety reports received over a 3-month period, starting 6 months before their involvement in our study. Investigators were then asked to document specific actions taken as a result of any report (eg, changes in informed consent; protocol inclusion and exclusion criteria; screening procedures) (see Online Supplement 1). They were also asked to record approaches other than individual expedited safety reports that sponsors used to communicate safety information, such as aggregate line listings, periodic summaries of expedited safety reports, and data monitoring committee reports.
Comparison of US and EC Expedited Reporting Systems
A different group of site investigators was recruited to compare resources required to process individual expedited safety reports to those required to process aggregated IND safety reports over a similar period. To facilitate this comparison, we sought investigators who were participating simultaneously in one trial that distributed individual expedited IND safety reports to investigators (according to US regulations in effect before March 28, 2011) and in a second trial that distributed safety information to investigators via quarterly aggregate reports, a potentially less burdensome approach used for trials under the jurisdiction of the EC 4 and permitted under a waiver from the FDA. The project team identified 2 pharmaceutical companies (Wyeth and GlaxoSmithKline) that had obtained such a waiver and enlisted their support in identifying and communicating with investigators.
A questionnaire developed by the project team (Online Supplement 2) was distributed to investigators by the pharmacovigilance groups of the respective pharmaceutical companies. Test articles (ie, investigational products) studied under the waiver are referred to as Test Article A (used in a transplantation study) and Test Article B (used in an oncology study). To compare the resources required for sites to process expedited safety reports over a 3-month period using the 2 different reporting systems, sites were asked to complete the questionnaire for the trial that used the European aggregated reporting system and for a “comparator” trial that used the then-current US system of expedited reports for individual cases.
Data Analysis
Continuous variables were summarized as medians (interquartile ranges [IQR]). Rates of changes in the study protocol and informed consent were calculated per 100 expedited safety reports reviewed.
We determined the total time reported for reviewing and handling each expedited safety report from the prospective documentation of personnel effort. Respondents categorized time spent by 3 groups of personnel—physicians; other health care personnel (HCP), such as RNs, LPNs, and PharmDs; and non-HCP, such as administrative or clerical staff. We determined the average proportion of total time spent reviewing expedited safety reports for each group. Based on input from industry, academia, and clinical research organizations, we estimated the cost of personnel time to be US$150 per hour for physicians, $60 per hour for other HCP, and $30 per hour for non-HCP, plus an additional 30% benefit burden for each group. The estimated total cost per expedited safety report was defined as the product of recorded hours spent reviewing each expedited safety report and the estimated hourly cost of the personnel reviewing them. In addition, 2 sensitivity analyses were performed to explore the effect of higher and lower proportions of physician time.
This investigation was determined by the IRBs of the Duke University School of Medicine and the FDA to be exempt from IRB review. The University of Pennsylvania IRB reviewed and approved the protocol implemented at its institution, which evaluated the time and resources required to process expedited safety reports from study sponsors and assessed ways in which study conduct was altered by those reports.
Results
Of the 375 investigators solicited to participate, 63 indicated an interest and were sent study forms to document resource requirements and the impact of expedited safety reports on trials. Response rates were much lower than expected: despite multiple prompts via e-mail, only 6 investigators (9.5%) returned completed forms. Of these, 5 returned both prospective and retrospective forms, while 1 returned only a retrospective form. Detailed information on sites that provided prospective data is reported in Table 1.
Information on the 5 participating sites prospectively documenting resource requirements.
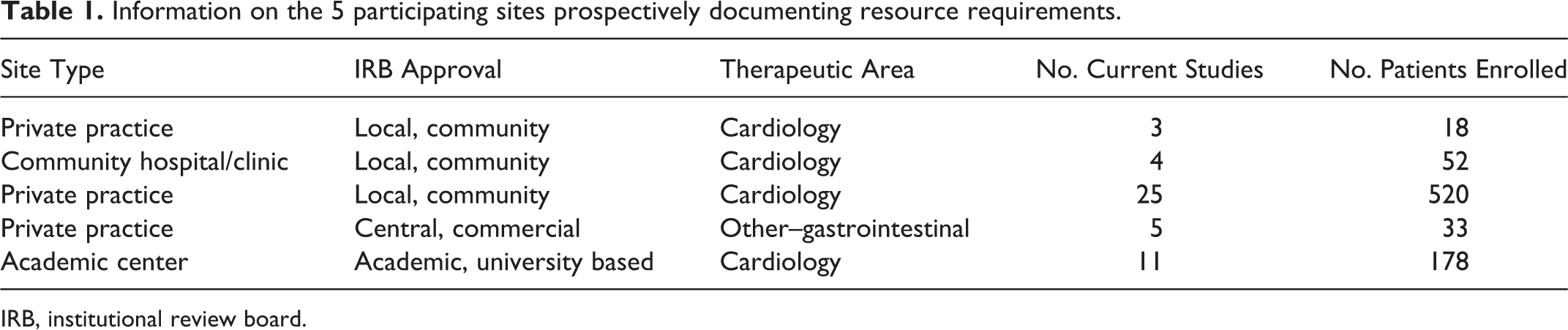
IRB, institutional review board.
For the comparison of allowed European and US reporting systems (ie, periodic aggregate reports via line listings vs individual expedited case reports), 46 site investigators were sent surveys for Test Article A, and 303 site investigators were sent surveys for Test Article B. Nine site investigators and their teams completed and returned data forms for each of the 2 test articles (18 total unique sites) for which there were FDA waivers allowing quarterly aggregate reporting.
Due to the very low response rate regarding resource requirements as well as the concentration of sites in a single therapeutic area (cardiology), our results, reported below, can only be considered anecdotal. In the Discussion section, we extrapolate our preliminary findings to 2 informative examples to exemplify the potential impact of our findings, if replicated, and detail a series of lessons learned from this project.
Resources Expended on Individual Expedited Safety Reports
Sites that prospectively collected data on expended resources reported receiving a median of 1.25 expedited safety reports per trial per week (IQR, 0.13-9.13; maximum, 11.13). A total of 472 expedited safety reports were documented across 15 clinical trials. The median number of hours devoted per report was estimated to be 0.25 (IQR, 0.12-0.37).
The typical proportions of personnel time reported were 0.25 for physicians, 0.20 for HCP, and 0.55 for non-HCP, leading to a cost estimate of US$86 per hour, or $22 for each expedited safety report, with a range of $10 to $32 (based on IQR of time spent per report).
Comparison of Resources Required for Processing Individual Expedited Safety Reports Versus Quarterly Aggregate Reports
The 9 responding sites for each test article chose comparator trials that were similar to trials of Test Articles A and B in total number of subjects and participating sites, length of trial, and exposure to study medication. During the period covered by the most recent aggregate report regarding the test article that received the waiver, similar numbers of serious adverse events were seen in the sites’ chosen comparator trials. Similar trends also were seen in resource use for both test articles compared with their site-specific comparator trials. Furthermore, site personnel estimated that similar amounts of time were required to read, interpret, and (if applicable) distribute an aggregate quarterly report, compared with amounts of time required for similar activities in a comparator trial for an expedited safety report of a single case.
Rate at Which Expedited IND Safety Reports Led to Objective Actions Taken
We calculated an overall rate at which an expedited safety report led to a change in study treatment or informed consent or to institution of a study amendment. Across the 1575 expedited safety reports from 35 trials reviewed as part of this study, the overall rate for any of these actions per 100 expedited safety reports did not exceed 1.
Other Safety Reporting Approaches Used
Investigators retrospectively documenting report burden and response were asked what other safety reporting approaches were used by sponsors to provide them with information. Other approaches were reported for 9 of 32 trials: 8 sponsors constituted data monitoring committees; 2 used aggregate line listings; 2 used periodic summaries of expedited safety reports; and 2 used “other” approaches (not specified).
Discussion
Our study was intended to provide an empirical basis for characterizing the time and personnel resources required to process expedited IND safety reports and to document any resultant alterations of study conduct at clinical sites for multicenter trials prior to implementation of the FDA’s final IND safety reporting rule. Unfortunately, the very low response rate to our survey severely limits our ability to draw inferences from the data gathered. This low rate may have been due to multiple factors, which we explore in greater detail below.
Extrapolation of Study Findings to Other Scenarios
Although our estimate of the time and cost required to process a single expedited safety report cannot be generalized due to the small number of responding sites, a disproportionate representation of cardiovascular studies, and the possibility of nonresponse bias, we think that it is still of interest to reflect on the implications if such an estimate were to be replicated in other contexts. We therefore applied the median findings from our sample to 2 clinical trial scenarios: 1 actual and 1 hypothetical.
Scenario 1: Cardiovascular Megatrial
Completed in 2009, the ROCKET AF Trial 5 included 1178 clinical sites in 45 countries that enrolled more than 14,000 patients. Investigators received 316 expedited safety reports derived from ROCKET AF data over the 2.5 years of the trial. During this same interval, ROCKET AF investigators received an additional 1134 expedited safety reports from other trials of the same investigational product and from postmarketing surveillance studies in countries that had already granted regulatory approval for the product (personal communication, Karen Hannan, project leader, Duke Clinical Research Institute, May 2012), for a total of 1450 reports over the course of the trial. Multiplying that number by 1178 investigators yields a total of 1,708,100 expedited safety reports delivered to investigators over 2.5 years. Applying our estimate of approximately 0.25 hours per report and median cost of personnel time of US$22 per report, the cumulative amount of investigator time spent processing these expedited reports over the course of the trial would be 427,025 hours, or 10,676 forty-hour workweeks, at an aggregate personnel cost to clinical sites of US$37.6 million. The magnitude of this estimate, which is generally consonant with estimates derived from our own experience (unpublished data, Bruce Burnett, PhD, Duke Translational Medicine Institute, 2008), underscores the importance of the FDA’s 2010 final IND safety rule with regard to decreasing the number of expedited reports that are not interpretable as individual events.
Scenario 2: Hypothetical Small Trial
We also applied our estimate to a hypothetical small trial. We used the median estimates from the 9 comparator trials of Test Article A that we had employed in evaluating differences between US and EC reporting regimes. A median of 302 subjects per trial were enrolled, and 50 sites were included per trial; subjects participated for 12 months, with a full 12 months of drug exposure. In those trials, a median of 10 expedited safety reports were received over 3 months, or 40 reports over the 12-month course of the trial. With 50 sites, a total of 2000 expedited safety reports would have been sent to investigators in these trials. Applying our estimate of time and cost per individual expedited report, such a trial would consume 500 hours (12.5 workweeks) at a cost of US$44,000 over the duration of the trial.
If these estimates were to be validated, our primary concern for the clinical trials enterprise would not be the monetary cost of these activities but rather the extent to which investigators and their staff are spending time on activities that may not add to their understanding of the risk-benefit balance of the investigational product. Time spent processing uninterpretable reports is time that cannot be devoted to carefully conducting the trial and ensuring the safety, rights, and privacy of trial participants.
The anecdotal complaints that prompted our study centered on the large volume of expedited IND safety reports that were purported to be uninterpretable as individual cases. Shortly after we completed our study, the FDA issued a final IND safety reporting rule that was intended to improve the utility of IND safety reporting by reducing the number of uninterpretable individual case reports, which would in turn reduce the burden of reviewing reports deemed unlikely to enhance the safety of trial participants. In an accompanying guidance for industry, the FDA commented that under the former IND safety reporting rule (21 CFR 312.32[a]), “sponsors frequently reported as individual cases serious adverse experiences for which there was little reason to believe the drug caused the event.” 6 The FDA noted that serious adverse experiences that are likely to be manifestations of the underlying disease, that are common in the population being studied (eg, strokes or myocardial infarctions in an elderly population), or that are study endpoints are frequently sent to the FDA and investigators as expedited IND safety reports but are “generally uninformative when reported as a single event (ie, without a comparison of the incidence of the event in treated and untreated subjects).” 6
Implications for Future Research
We note that the low response rate to our study constitutes a finding of some importance in itself. Earlier anecdotal communications and widespread perceptions in the field of clinical research indicated that the burden imposed by the existing expedited event reporting system constituted a serious problem for clinical trials personnel, as well as a potential detriment to the efficient conduct of research and to ensuring the safety and well-being of trial participants. Furthermore, even in the wake of changes to reporting rules, there is still concern that sponsors’ practices have not changed and that significant numbers of uninterpretable expedited case safety reports continue to burden the system. 7
If we are to make evidence-based regulatory improvements and protect patient safety, it remains critical that changes to existing regulatory requirements be guided by objective, generalizable evidence about their effectiveness. However, the relative dearth of systematic investigation in this arena and the disappointingly low response rate to our own study suggest that serious thought should be given to optimizing approaches to this type of research. Below, we summarize 2 key lessons derived from our experience.
Research on the Effects of Regulations Must Minimize Imposition of Additional Burdens Upon Sites
Although prospective recording of resources devoted to processing safety reports was needed to avoid recall bias in our study, the paper forms used for this purpose may have constituted an excessive burden for investigators and site personnel. It is possible that an electronic data capture system that prioritizes ease of use might alleviate this problem, at least in part. A survey of site attitudes toward large simple trials conducted by RTI International (http://www.rti.org/; Research Triangle Park, North Carolina, USA) achieved much higher rates of response (53 of 89, 60%) using customized web modules to capture information. 8 The ultimate method for such data acquisition would be an electronic health record optimized for the learning health care environment that automatically captures information that can be used for both clinical research and quality improvement initiatives, 9 including assessing the impact of regulatory requirements. Pilot programs that leverage electronic health records for research data collection within the practice environment are already underway through the National Institutes of Health’s Health Care Systems Research Collaboratory (https://www.nihcollaboratory.org/demonstration-projects/Pages/default.aspx); expanding such capabilities to inform regulatory science would be a logical extension of these efforts.
Stakeholder Involvement and Engagement Is Crucial to Success
Another factor that we think contributed strongly to the successful survey investigation described above was early and significant engagement of potential survey participants, who were invited to take part in a “think tank” discussion that informed the study design and implementation. We note that in addition to the resource and workload burdens imposed by the clinical trials setting, many clinicians lack incentives to take part in clinical research 10 ; indeed, many investigators may face de facto penalties for such activities due to institutional or health system emphasis on operational efficiency and other “bottom-line” priorities. 11,12 Given this background, we believe that it is essential to provide potential participants with meaningful incentives for engaging in “research about research,” and a critical aspect of this process is soliciting input on study design and implementation methods that will ensure adequate response rates and high-quality actionable data.
The only way to be sure of the impact of new regulations is to document their effects by gathering reliable empirical evidence that can inform research. High-quality information on the outcome of well-intended regulation will enrich the scientific basis for future actions; however, acquiring such information requires thoughtful approaches to research design, active stakeholder engagement, and meaningful commitment from sponsors, investigators, and regulators.
Footnotes
Authors’ Note
The views expressed herein represent those of the authors and do not necessarily represent the views or practices of the authors' employers or any other party.
Acknowledgments
The authors thank Jonathan McCall, MS, and Amanda McMillan, MA, MPH, of the Duke Clinical Research Institute, Durham, NC, USA, for editorial assistance. Neither Mr. McCall nor Ms. McMillan received any compensation for their work other than their usual salaries.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was conducted through in-kind contribution of effort by authors and organizations working with the Clinical Trials Transformation Initiative (CTTI; ![]() ), as well as staff effort and meetings supported by CTTI funding (derived both from pooled membership fees of member organizations and from cooperative agreement U19FD003800 awarded to Duke University from the U.S. Food and Drug Administration). Amgen contributed a part-time project manager supported by a CTTI staff member. ICON contributed programming and data management services supplemented by a CTTI staff member. ICON, Duke Clinical Research Institute, and the University of Pennsylvania all contributed to the statistical analysis.
), as well as staff effort and meetings supported by CTTI funding (derived both from pooled membership fees of member organizations and from cooperative agreement U19FD003800 awarded to Duke University from the U.S. Food and Drug Administration). Amgen contributed a part-time project manager supported by a CTTI staff member. ICON contributed programming and data management services supplemented by a CTTI staff member. ICON, Duke Clinical Research Institute, and the University of Pennsylvania all contributed to the statistical analysis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.