
Abstract
The systems theory of autistogenesis accounts for genetic and environmental predisposing factors for pervasive developmental disorders. During development, regions of the brain myelinate differentially, even while neuroinflammatory events induce neurological damage. Incorrect dietary ratios of docosahexaenoic acid (DHA) to arachidonic acid (AA) promote developmental aberration characteristic of autism spectrum disorders (ASD), and commercial infant formulae possesses DHA/AA ratios unsuitable for normal brain development in those predisposed. The aromatase gene regulates DHA/AA metabolism and represents a potential biomarker for ASD. Aromatase converts testosterone to estradiol. Estradiol is neuroprotective and a modulator of oxytocin receptors deficient in autism. Neuroprotective DHA is not well synthesized in males and is regulated by estradiol. Therefore, converging evidence indicates that any disturbance to the autistogenic system linking environment to neurobiology and genetics is capable of inducing developmental disorders with gender disparity.
Autism spectrum disorders (ASD) are characterized by signs that may be detectable by 6 months of age (Brooks & Meltzoff, 2007, 2008; Meltzoff & Brooks, 2007; Roos, McDuffie, Weismer, & Gernsbacher, 2008) that include (a) qualitative and quantitative impairments of social interaction, (b) deficits in the use and production of communicative means, (c) cyclic central nervous system (CNS) activity producing repetitive behaviors, and (d) a preference for environmental sameness coupled to an aversion for extremes in sensory novelty (Belva, Matson, Hattier, Kozlowski, & Bamburg, 2012; Lundström et al., 2012). The number of children diagnosed with disorders on the autism spectrum has increased since 2001 (Bertrand et al., 2001; Chakrabarti & Fombonne, 2001) to an approximated 3.5 million children nationwide, or an estimated 1 out of every 91 individuals in the United States (Kogan, Blumberg, Schieve, Boyle, & van Dyck, 2009) at a national cost of approximately US$26 billion annually (Baird et al., 2006) with males outnumbering females 4 to 1 (Fombonne, 2005). This degree of prevalence impels the search for discernible etiology, biological assay, evidence-based treatment, and practical routes to prevention.
Following Kanner (1943, 1949) and Bettelheim (1967), social outcry pushed many psychologists into a corner. While behaviorism was proclaiming mental disorder principally due to environment, parents argued any consideration of high-stress environments as attacking mothers, and many psychologists withdrew. Today, some still point to video data from Baranek (1999) suggesting that some parents may increase efforts to engage children on average by month 13.86 (SD = 7.43) though diagnosed with autism on average by month 32.55 (SD = 8.62). This study is sometimes taken to “prove” parental efficacy, although current studies (Brooks & Meltzoff, 2007, 2008; Meltzoff & Brooks, 2007; Roos et al., 2008) indicate that awareness of developmental abnormality likely began earlier than the stated time for parents’ first detection, standard deviation included (Baranek, 1999). This does not necessarily suggest attempts at deception. Neurotypical caregivers discern changes and respond in typical fashion, but these videos do not and cannot describe the early environment, in a broad sense, in relation to genetic variability prior to detection.
Neuropsychologists, neurophysiologists, psychiatrists, endocrinologists, immunologists, and behavioral geneticists continued to study the environmental influence on neurodevelopment, including that of infant maltreatment. For example, in the largest and most recent twin study of autism (Hallmayer et al., 2011), following current diagnostic standards, monozygotic and dizygotic pairs with at least one twin clinically diagnosed with an ASD or strict autism, environment was the critical factor overall. Heritability was estimated at .38, 95% confidence interval (CI) [.14, .67] for those with an ASD, whereas the shared environment was .58, 95% CI [.30, .80] (Hallmayer et al., 2011) regardless of gender. For those diagnosed with strict autism, males and females were again the same, where heritability was estimated at .37, 95% CI [.08, .84], whereas the shared environmental aspect was .55, 95% CI [.09, .81] (Hallmayer et al., 2011). This article does not suggest that all ASD is the result of widespread child abuse. Maternal nutrition and stress-induced epigenetic DNA regulation and methylation, neonatal nutrition, the general stress surrounding the home (e.g., urban areas of high violent crime, regions of the globe shattered by war, infectious disease), and environmental toxins are also part of the environment, and the common impacts on biology are key.
The purpose of this work is to synthesize from the available information a framework on which to build; to report that model as a unified whole, at one time; and through the best medium available, to avoid sequestration within disparate specialty journals. The result is the systems theory approach to autistogenesis (Malone, 2011a, 2011b, 2011c, 2011d), unifying current findings from diverse fields, capable of explaining why the previous study (Hallmayer et al., 2011) also detected a range of heritability (41% to 56% in males, 13% to 16% in females), yet shared twin environment (57% to 41% in males, 78% to 72% in females; Hallmayer et al., 2011).
This work begins with an examination of environmental stressors on infant neuronal development. Second, a source of dysfunction not well described is the potential hazards resulting from the metabolism of inadequate docosahexaenoic acid (DHA; n-3) to arachidonic acid (AA; n-6) ratios in breast milk and infant formula. Third, the influence of cytochrome P450-dependent and enzyme-independent reactions, which may convert DHA and AA into neuroprotective agents or toxic proinflammatory metabolites, are examined. Finally, this work identifies the predisposing gene that explains vulnerability to environmental triggers, reveals nearly a dozen links of male overrepresentation, and why both disintegration disorder and posttraumatic stress disorder (PTSD) are causally linked to the same pathophysiological processes.
Method
Inclusion–Exclusion Criteria
Evidence-based medicine often relies on metanarrative review, and this method is used to illuminate how biology and environment intermingle in the systems theory of autistogenesis (Malone, 2011a, 2011b, 2011c, 2011d). Given that several journals are dedicated to autism research, and that many journals continually publish additional studies bearing on this multifaceted developmental disorder, inclusion criteria were necessarily liberal. Particular areas reviewed included clinical nutrition, pediatric neurology, lipid metabolism, cognitive neuroscience, molecular and cellular biology, clinical psychology, pharmacology, neurophysiology, developmental psychology, endocrinology, archeology and biological anthropology, brain morphology, immunology, genetics, and toxicology. Journal articles, conference papers and presentations, books and book chapters, and dissertations were considered eligible for inclusion. The nature of this study required direct telephone contact with pharmaceutical companies to ascertain chemical constituents. Resources not addressing associated genetic or metabolic processes, pervasive developmental disorders, autistic behaviors, or the evolutionary significance of such processes were typically excluded.
Literature Search
Library holdings, MEDLINE, PsycINFO, ERIC, Academic Search Complete, JSTORE, ProQuest Central, SAGE Online Journals, Google Scholar, and the eBAY and PsycBOOKS e-book collections were explored to obtain articles for the present study. Studies containing permutations of the words autism, developmental disorder, infant, genetic linkage, mirror neuron, docosahexaenoic acid/DHA, fetal, arachidonic acid/AA, inflammation, sensitive period, environment, androgen, testosterone, estrogen, estradiol, fish oil, milk formula, stress, breast milk, and gender were considered. Reference lists of all acquired articles and book chapters were reviewed for potential sources, with allowances made for older works where previous findings illuminate current research in dissimilar fields of study.
Results
Sensitive Periods
Penfield and Roberts (1959) first described a sensitive period in language acquisition, and since Lennenberg (1967/1984), continuing research demonstrates that production, use, and understanding of a primary communicative means is ontogenically constrained. Restriction of competence, performance, and perception is confirmed through studies of the home environment (Sohr-Preston & Scaramella, 2006), examination of language-deprived children (Curtiss, 1977, 1989), functional magnetic resonance imaging (fMRI) studies of language lateralization and localization (Ahmad, Balsamo, Sachs, Xu, & Gaillard, 2003; Fogassi & Ferrari, 2007; Gaillard, Balsamo, Ibrahim, & Xu, 2003; Sachs & Gaillard, 2003), and both direct cortical stimulation and Wada testing (Schevon et al., 2007). While it is true that second languages may be learned at a later time, it is also true that the process of second-language development occurs through a neurologically different process ever after (Friederici, 2009; Pinel & Dehaene, 2010; Ylinen et al., 2010), and language is only a portion of the broader realm of developmental sensitivity.
Sensitive periods exist throughout the brain due to substantial synaptic remodeling of cortical connections (Finlay & Slattery, 1983), many becoming “hard-wired” through myelination, diminishing plasticity (Giedd et al., 1996; Gogtay et al., 2004; Qin et al., 2004; Sowell et al., 1999), due in part to inhibitory myelin-dependent signaling (Puttaguntal et al., 2011) and membrane-bound myelin-associated glycoproteins (MAGs) that inhibit axonal growth and synaptogenesis (Llorens, Gil, & del Rio, 2011). The result is broad pathways modification governing development of perception (Kalia, Legge, & Giudice, 2008), shared attention (Brooks & Meltzoff, 2008), learning, memory, language, rudimentary mathematics, and social engagement (Allman, Watson, Tetreault, & Hakeem, 2005; Bijeljac-Babic, Nassurally, Havy, & Nazzi, 2009; Butterworth & Morisette, 1996; Camaioni, Castelli, Longobardi, & Volterra, 1991; Greenfield, 2000; Greenfield, Maynard, Boehm, & Yut, 2000; Iacoboni, 2009; Koechlin, Dehaene, & Mehler, 1997; Wynn, 1992, 1995).
The brain adapts to coconstruction of meaning soon after birth (Gao, Levine, & Huttenlocher, 2000; Kuhl & Meltzoff, 1996; Starkey, 1992) as attachment secures enculturation (Ainsworth, Blehar, Waters, & Wall, 1978; Bowlby 1969, 1988). Because mirror neurons permeate the cerebral cortex (Mukamel, Ekstrom, Kaplan, Iacoboni, & Fried, 2010) in regions maturing differentially, this work asserts that the existence of sensitive periods for aspects of mimicry, imitation, goal emulation, and reciprocity are certain.
Stressors and the Sensitive Period
In patients with autism, cerebellar folia Purkinje cells (PC) approach 15 standard deviations below the mean compared with controls due to neuroinflammatory processes (Kern & Jones, 2006). Immature PCs receive presynaptic inputs from olivocerebellar cells and experience axonal elongation through search mechanisms to deep nuclear neurons and other postsynaptic output targets (Dusart, Airaksinen, & Sotelo, 1997). The output from the cerebellar cortex are PCs, which inhibit several regions of the cerebral cortex and dendritic development connecting to olivary cells, and require high cerebellar estradiol (Figure 1:4; Figure 4; Figure 6:1; Keller, Panteri, & Biamonte, 2010). Increased microglial and astroglial activation associated with the neuroinflammatory response (Figure 2:1) is found in areas of reduced PCs, astroglial response in the granular cell layer of the cerebellar white matter, and the middle frontal gyrus (MFG) and anterior cingulate gyrus (ACG) of autistic patients (Vargas, Nascimbene, Krisnan, Zimmerman, & Pardo, 2004). The reduction in PC development is male biased and linked to a gender-based mechanism described in this study.
Neuroinflammation resulting from insult or injury, including apoptosis resulting from chronic or repeatedly acute cortisol exposure (Montuschi, Barnes, & Roberts, 2004; Uz et al., 1999), up-regulates neurotoxic inflammation mediators (Figure 2:2; Block & Hong, 2005; Floyd, 1999). These processes result in autotoxic cycles leading to further neurodegeneration (Licastro et al., 2005), and autistic changes are not restricted to olivary-cerebellar pathways. Von Economo neurons (VEN) project from the anterior cingulate cortex (ACC) and extend through Layer 6 of the frontoinsular cortex (FI) and to prefrontal, orbitofrontal, insular and anterior temporal cortices, the amygdala, hypothalamus, thalamic nuclei, and periaqueductal gray matter (Allman et al., 2005). Individuals on the autism spectrum demonstrate significantly reduced, disordered, and dysfunctional VEN with associated migration deficits. (Allman et al., 2005).
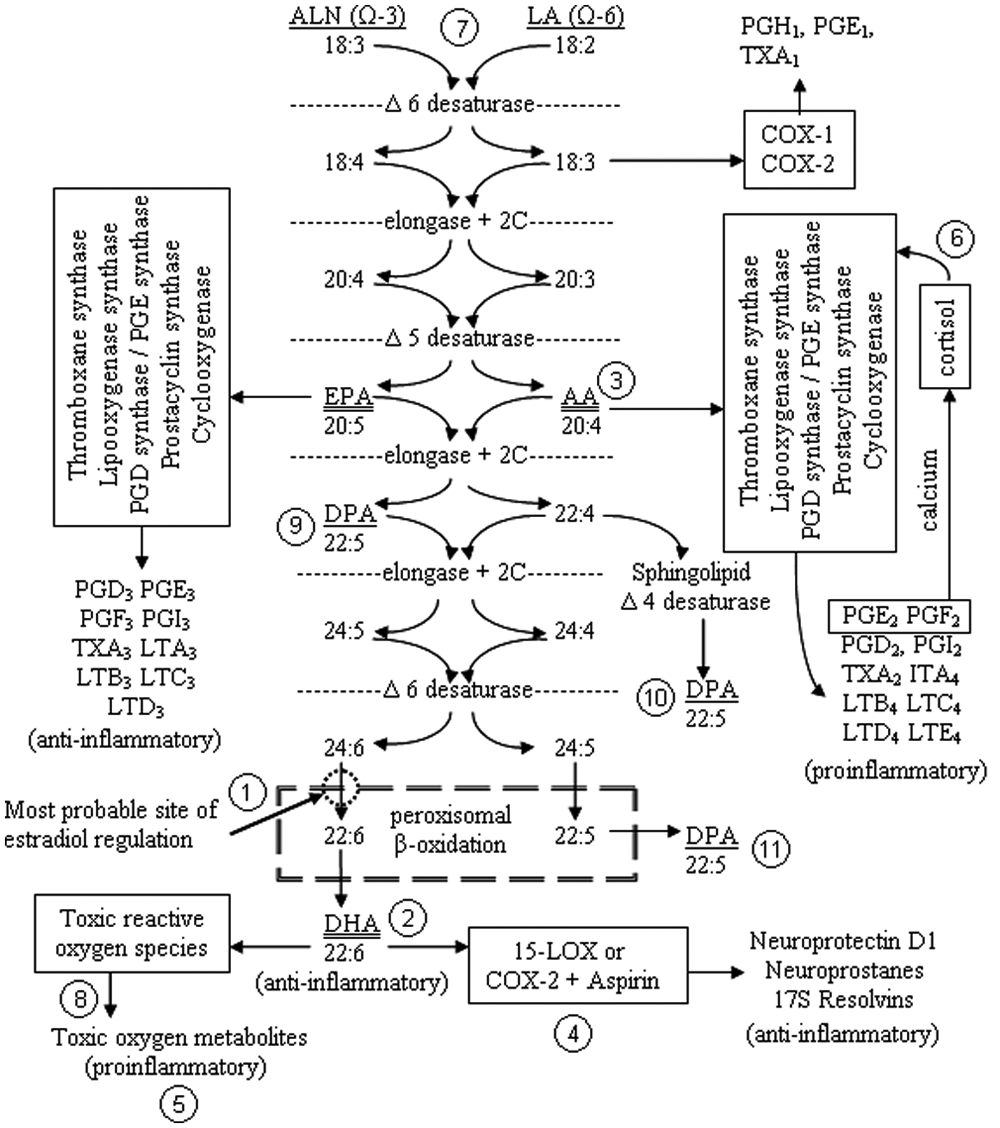
Dietary ALN is transformed into EPA through the same pathway that LA is converted into AA. Following elongation and desaturation, there are three routes to producing DPA, but there appears only one pathway from precursors to DHA that can be ultimately converted to NPD1
Chronic neuroinflammation of similar etiology during infancy will produce strikingly similar effects if circumstances occur during the same developmental periods (Rutter et al., 1999). The mirror neuron system (MNS), well rooted in the ventral premotor cortex, and the anterior, inferior fronto- and tempero-parietal regions, permeates the cerebral cortex (Mukamel et al., 2010), including the primary somatosensory cortex. The MNS is intensely interconnected with limbic system, orbital frontal lobes, and insula, and each region experiences myelination at different rates during different periods of time (Huttenlocher, 1990; Yakovlev & Lecours, 1967).
Deficits within the MNS represent a key neurological correlate to quiescent social behaviors diagnostic of autism (Iacoboni & Dapretto, 2006; Schmitz & Rezaie, 2008; Williams et al., 2006; Williams, Whiten, Suddendorf, & Perrett, 2001). MNS activates while witnessing another’s actions, producing matching somatosensory and proprioceptive feedback mapped onto the self (Fogassi & Ferrari, 2007; Rizzolatti & Destro, 2007). The MNS is modulated by emotion and motivation, provides the access to empathic experience of another, and creates the means by which facial expressions and other communicative gestures are interpreted (Bufalari, Aprile, Avenanti, Di Russo, & Aglioti, 2007; Cheng, Meltzoff, & Decety, 2007; Iacoboni, 2007; Iacoboni & Dapretto, 2006; Meltzoff, 2008; Schmitz & Rezaie, 2008; Williams et al., 2006), and like VENs, it is highly susceptible to neuroinflammatory damage. Because the MNS, in conjunction with the limbic system, experiences myelination at various rates in many brain regions, high stress environments during different developmental stages may induce abnormal interconnectivity similar to the effects of androgen-induced cytotoxicity (Blaylock, 2009).

The neuroinflammatory cascade results from illness or injury, whether physical or psychological, and leads to the removal of dead, infected, damaged, or physiologically impaired cells within the brain. Initiation of neuroinflammation is a feed-forward process, may persist from 10 days to 2 weeks, and can damage or destroy progressively expanding layers of cells until resolution
That developing under the stress of warfare or intense urban violence may result in childhood behavioral abnormalities should not be a surprise. Hunt (1941), Kanner (1949), and Spitz (1945) suggested that behaviors characteristic of ASD may be induced through abusive rejection and social isolation during infancy (Bettelheim, 1967; Lundin, 1971). Many have confirmed these observations in Russian orphanages, Romanian foundling homes (Gunnar, Morison, Chisholm, & Schuder, 2001; O’Connor & Rutter, 2000; Rutter et al., 1999; Smith, Greenberg, Seltzer, & Hong, 2008; Tustin, 1991, 1994), and modern social pediatrics (Hellbrügge, 2010; Hellbrügge, von Wimpffe, Lajosi, Menara, & von Sarközy-Kerner, 1973; Schneeweiß & Hellbrügge, 2010). Recently, Hane, Henderson, Reeb-Sutherland, and Fox (2010) completed a longitudinal study indicating that low-quality caregiving within the realm of ordinary variation is sufficient to modify social development and frontal electroencephalogram (EEG) asymmetry in young children, with male bias. When the socially sensitive limbic system receives inappropriate or inadequate stimulation, synapses undergo apoptosis, while abnormal connections myelinate (Bremner et al., 1995; Gähwiler, 1983; Stein, Hannah, Koverloa, & McClarty, 1995).
Research indicates that maltreated children can develop posttraumatic stress disorder–like symptoms (Kearney, Wechsler, Kaur, & Lemos-Miller, 2010) comorbid with significantly reduced corpus callosum in males (De Bellis et al., 1999; De Bellis & Kuchibhatla, 2006) and demonstrates that caregiver maltreatment of young children is linked to later bullying (Hong, Espelage, Grogan-Kaylor, & Allen-Meares, 2011). Male infants chronically exposed to physical abuse may develop reduced language competence, diminished social awareness, lack of attention, minimal empathic response, and uninhibited and ritualized aggression, whereas females may not due to reduction in activation of stress response pathways (Goldstein, Jerram, Abbs, Whitfield-Gabrieli, Makris, 2010). Maternal stress during pregnancy (Wright & Enlow, 2008) and abuse in infancy is linked to increased DNA methylation within the hippocampus (McGowan et al., 2009) and an approximately threefold increased methylation of the brain-derived neurotrophic factor gene (BDNF) in the prefrontal cortex (Roth, Lubin, Funk, & Sweatt, 2009).
Maltreatment and abusive rejection initiates the neuroinflammatory cascade (Figure 2:3; Figure 6:2; Slavich, Way, Eisenberger, & Taylor, 2011) leading to diminishment of connections to and from the amygdala, septal nuclei, cingulate gyrus, hippocampus, and the neocortex as neurite formation and synapses per axon are reduced, with retention of unsuitable interconnections resulting as requisite apoptosis fails due to continuing myelination (Joseph 1982, 1992, 1996). Another study (Danese et al., 2010) followed children from 41 homes participating in the Risk Longitudinal Twin Study (Moffitt & the E-Risk Study Team, 2002), comparing physically maltreated children against those from homes where no maltreatment was detected. The results demonstrate that children scoring higher than the validated stop point (CDIX20) on the Children’s Depression Inventory (Kovacs, 1992) had significantly increased proinflammatory metabolites, geometric mean = 0.49, p = .016, 95% CI [.28, .84], with male bias (Danese et al., 2010).

A comparison of American breast milk DHA/AA ratios as compared with countries surveyed. Source: Developed from data in Brenna et al., 2007. Copyright, 2007 by the American Society for Nutrition

Transcripts for tissue specificity must unite with enzyme expression and so increases the potential for dysfunction beyond mutation or transcript-level regulation of a single gene
These well-documented neurophysiological processes (Dudley, Li, Kobor, Kippin, & Bredy, 2011; Toyokawa, Uddin, Koenen, & Galea, 2012) may not happen at some predetermined developmental stage, all at once, or to the same degree. However, the results influence socially sensitive regions of the brain, and changes proceed in a fashion predicted by studies of learning and neuronal regeneration (Diamond, 1985, 1991; Greenough & Black, 1992; Greenough & Chang, 1988; Greenough & Volkmar, 1973; Walsh, Budtz-Olsen, Penny, & Cummins, 1969). Because different aspects of developmental sensitivity arise differentially, it becomes immediately apparent how some individuals may possess fewer deficits in one domain while not in another, and demonstrates how plasticity may then advance development with gross asynchrony (Figure 7).
Malformed neurodevelopment occurs in high-stress, neglectful, or abusive environments as the neurology maladapts in a hostile environment (Andersen et al., 2008; Gatzke-Kopp, 2011; Hoftman, & Lewis, 2011; Lupiena et al., 2011; Shonkoff, 2011; Walker et al., 2011). Enhanced short-distance connections create a blockade to further long-distance neuronal organization (Courchesne et al., 2007), and interconnectivity is nullified as cortisol and cytotoxic inflammatory metabolites degrade fledgling connections (de Quervain, Roozendaal, & McGaugh, 1998; de Quervain, Roozendaal, Nitsch, McGaugh, & Hock, 2000; Sapolsky, 1992). The results of these processes represent a likely component to childhood disintegration disorder (Malone, 2011d), originally a diagnostic sign of classical autism in video studies (Baranek, 1999).

The horizontal reference lines indicate logarithm of odds score = 2.2 suggestive of linkage, and 3.6 = statistically significant for ASD. Genetic location over 50 indicates traits expressed on other chromosomes. Both IQ ≥ 70 and restricted, repetitive, and stereotyped behaviors (BEH) demonstrate positive linkage

Autism spectrum and other developmental disorders are the result of damage to or impairment of one or more components within a system of interconnected processes; the brain’s multidomain periods of developmental sensitivity, the influence of internal and external environmental stressors on developing neurology, sex hormone regulation of LC-PUFA metabolism, and the genetic mechanisms and metabolic processes of testosterone conversion to estradiol
Corrupt neurite formation and neural migration in autism yields cortical dysgenesis, including cortical thickening, fewer interconnections, increased density with intermingling gray and white matter, and protruding gray matter beyond outer layers (Schmitz & Rezaie, 2008). Early chronic or repeatedly acute stress may produce neuroinflammation and induces similar effects in mammalian models studied, whether dogs, nonhuman primates (Geyer, Wilkinson, Humby, & Robbins, 1993; Harlow, 1974; Harlow & Harlow, 1965; Kling, 1972), or even human children (Goldfarb, 1943, 1945; Spitz, 1945, 1946). These factors contribute to the development of ASD typical behaviors, including (a) diminished perceptual, intellectual, and learning capacities, (b) inability to inhibit repetitive or self-destructive behaviors, and (c) gross distortion of communicative, emotional, and social operations.

The postnatal prepubescent sex hormone surge occurs just prior to the onset of most developmentally sensitive periods (for 23 specific developmentally sensitive periods assessed, see van Os, Kenis, and Rutten, 2010)
DHA as an Essential Fatty Acid
DHA is critical for the production and efficient function of neurons. Although not typically considered an essential fatty acid because synthesis occurs in humans, DHA requires several steps (Figure 1:2; Figure 6:3) toward elongation and desaturation from α-linolenic acid (ALN; 18:3 n-3). Considered ubiquitous in neuronal tissues, the cerebrovascular endothelium and astrocytes provide active transport, and only the liver and some astrocytes are capable of significant DHA synthesis (Moore, 2001). There is clear in vivo evidence that DHA is vitally important to learning and memory at the component level as deficiency studies reveal smaller neuronal cell bodies in the hippocampus, hypothalamus, and the parietal cortex (Ahmad, Moriguchi, & Salem, 2002). Uptake is highest during the final trimester, which also represents the greatest phase of neurogenesis, neurite formation, and arborization (Green & Yavin, 1998).
Judge, Harel, and Lammi-Keefe (2007) detected significant positive effects of DHA supplementation on problem solving by 9 months of age. The neural correlates to DHA’s effects are clear; size and quantity of neurite formation are promoted by DHA while inhibiting apoptosis induced by sphingosine (Hashimoto et al., 2005; Ikemoto, Kobayashi, Watanabe, & Okuyama, 1997; Okada et al., 1996), cultivating neuronal differentiation, exciting neurite growth factors (Calderon & Kim, 2004; Kawakita, Hashimoto, & Shido, 2006), and enhancing phospholipid manufacture required for neurite elongation (Horrocks & Yeo, 1999; Kan, Melamed, Offen, & Green, 2007). Following the same enzymatic pathway as DHA (Figure 1:3; Figure 6:4), AA is synthesized from linoleic acid (LA; 18:2 n-6). Neurite outgrowth is suppressed by AA (Ikemoto et al., 1997) through the inhibition of ethanolamine glycerophospholipids, the induction of the inflammatory cascade (Figure 2:4), and in concert with tumor necrosis factor (Wissing, Mouritzen, Egeblad, Poirier, & Jaeaettelae, 1997).
Studies indicate that long-chain polyunsaturated fatty acids (LC-PUFAs) are critical to neurodevelopment and function, including their connection to neuropsychological disorders (Boure, 2004; Haag, 2003; Jefferey, Waisinger, Nauringer, & Mitchell, 2001; Mitchell, Niu, & Litman, 2003). The following sections describe the interplay between DHA, AA, and other LC-PUFAs, and the metabolic responses to stressors capable of inducing and mitigating neuroinflammatory responses. This work then introduces the genetic mechanism responsible for several routes to male overrepresentation and illustrates how environmental triggers potentiate biological processes linked to LC-PUFA metabolism in autistogenesis.
Lipid Metabolism and Stress
Because DHA is shunted through the phosphatidylserine (PS) synthesis pathway, translocation of Akt and Raf-1 protein kinase genes enhance cell survival when phosphatidylinositol-3,4,5-triphosphate (PIP3) activation of Akt is reduced or neuronal differentiation is required (Akbar, Calderon, Wen, & Kim, 2005; Kim, 2007; Kim, Akbar, Lau, & Edsall, 2000). DHA demonstrates a strong influence on Na+, K+, and Ca+ (Figure 1:6; Figure 2:3) channels, promotes synaptic transmission, and enhances long-term potentiation associated with learning (Itokazu, Ikegaya, Nishikawa, & Matsuki, 2000; Poling, Vicini, Rogawski, & Salem, 1996; Vreugdenhil et al., 1996; Young, Gean, Chiou, & Shen, 2000; Young, Gean, Wu, Lin, & Shen, 1998). DHA demonstrates antiapoptotic effects by increasing PS synthesis and diminishes capase-3 activity, a mediator of apoptosis, at the level of transcription (Kim, Akbar, & Kim, 2001; Kim et al., 2000). Furthermore, DHA up-regulates the antiapoptotic Bcl-xl, Bcl-2, and Bfl-1(A1) gene expression while down-regulating proapoptotic Bax and Bik expression (Lukiw et al., 2005).
DHA is also transformed by an enzyme similar to 15-LOX (Figure 1:4) into neuroprotectin D1 (NPD1), which is promoted during return of blood to tissues following ischemia, suppresses Aβ-42-induced toxicity in Alzheimer disease, inhibits apoptosis, blocks leukocyte infiltration, and modulates proinflammatory gene expression (Horrocks & Farooqui, 2004; Morris et al, 2003). The inflammation cascade from AA metabolism is inhibited by DHA that enhances cell membrane fluidity, modulates protein kinase activity, inhibits membrane surface antigens and adhesion molecules induced by cytokine expression, and suppresses macrophage nitric oxide synthase (Takahashi et al., 1997).
Free DHA is regulated in the brain as oxidative stress produces reactive oxygen species capable of generating DHA peroxidation products (Figure 1:5) that induce cell damage (Mukherjee, Marcheselli, Serhan, & Bazan, 2004). Under great stress, cortisol induces enhanced lipoxygenase expression and free radical-mediated peroxidation, which creates proinflammatory prostaglandins from AA and DHA (Figure 1:6; Figure 2:5; Montuschi et al., 2004; Uz et al., 1999). Inflammatory metabolites of AA also include leukotrienes (Figure 2:6), which sustain the inflammatory response, and hydroxyeicosatetranoic acids which are released while conversion of DHA into resolvins and protectins by a 15- lipoxygenase-like enzyme self-limits the response (Farooqui, Horrocks, & Farooqui, 2007; Horrocks & Farooqui, 2004; Lukiw et al., 2005).
Chronic neuroinflammation may lead to nonenzymatic oxidation of DHA producing reactive neuroketals and generates further toxic metabolites (Figure 6:5; for a list of proinflammatory AA and DHA biomarkers, see Farooqui et al., 2007). These processes also occur when eicosapentaenoic acid (EPA) is added without sufficient DHA (Carlson, Werkman, & Tolley, 1996), and the potential negative effects of adding either without antioxidants (Halliwell & Chirico, 1993). Diep, Touyz, and Schiffrin (2000) indicate that nonenzymatic oxidation of DHA may induce apoptosis through a translocation of PS, heightened Bax, and capase-3 activity subsequent to disruption of mitochondrial transmembrane potential and activation of peroxisome proliferator-activated receptors (PPARs) via the p38 signaling pathway.
AA is made throughout the body and stored within cell membranes of every tissue as a reservoir for the inflammatory response. Inflammation begins when cell disruption (Figure 2:7) induces the enzymatic conversion of free AA (arachidonate) into lipoxygenase-derived leukotrienes and cyclooxygenase-derived prostaglandins, and thromboxanes, each of which may result in additional toxic oxygen metabolites (TOM; Figure 2:8; Becker, Reece, & Poenie, 1996; Berg, Tymoczko, Stryer, 2010). Eicosanoids produced from this series of events not only alter vascular permeability (Figure 2:9) resulting in tissue swelling, but also lead to the production of more TOM while the apoptosis chain-reaction causes the release of more than 50 lysosomal enzymes (Figure 2:10) that injure or destroy neighboring cells, and (Becker et al., 1996; Berg et al., 2010) lysosomal enzymes also cause cytokines (Figure 2:11) to activate complement (Figure 2:12), leading to further changes in vascular permeability, and initiating leukocytosis (Figure 2:13; Rhoades & Planzer, 1996).
Cellular disruption also induces brain mast cell degranulation (Figure 2:14), which results in a release of histamine, serotonin, and heparin (Figure 2:15; Dvorak et al., 1992; Johnson & Krenger, 1992; Marshall, 2004; Penner & Neher, 1989). Some AA metabolites are capable of anti-inflammatory actions (Figure 6:6; Node et al., 1999), although the effect of AA on the overall self-limiting reaction may not diminish the inflammatory response at the tissue level for more than a week in humans. Therefore, a careful balance must be struck between AA and DHA as both share the same desaturation and elongation pathway (Figure 1:7) when converted from LA and ALN, respectfully.
Epinephrine is also released during acute moments of stress (Figure 2:16). Repeatedly acute periods of stress alter monoamine oxidase A (MAO-A) activity (Figure 2:17) on serotonin, epinephrine, norepinephrine, dopamine, and phenylethylamine, and leads to further accumulation of TOM. Furthermore, the expression of this enzyme is regulated by the SRY gene responsible for male sex determination (Wu, Chen, Li, Lau, & Shih, 2009). A detailed flow diagram (Figure 2) illustrates the feed-forward inflammatory cascade, which results in the removal of injured or impaired brain cells (debridement) through autolysis, apoptosis, necrosis, and the action of brain macrophages and microglial cells (Figure 2:18; Fischer & Reichmann, 2001; Fujita, Yoshimine, Maruno, & Hayakawa, 1998; Hanisch & Kettenmann, 2007; Nimmerjahn, Kirchhoff, & Helmchen, 2005; Nottet, 1999; Tremblay et al., 2011). Brain macrophages and microglial cells engulf weakened cells or cellular debris and produce antibodies against similar cells and cellular components in essentially a localized autoimmune response (for a recent list of most known autoantibodies detected in patients with autism, see Torres, Westover, & Rosenspire, 2012).
Dietary LC-PUFA Supplementation Through Breast Milk and Infant Formula
That dietary intake (Figure 6:7) influences microsomal reactions in LC-PUFA biosynthesis is clear (Voss, Reinhart, Sankarappa, & Sprecher, 1991), and loss of DHA from adult healthy brain tissues is known to induce an inability to concentrate, loss of memory, and dementia (Pawlosky & Salem, 1995). This assertion is supported by a study of polymorphisms of delta-6 desaturase (Figure 1: 7, 10), necessary for conversion of precursors into AA, n-6 docosapentaenoic acid (DPA), the estradiol-dependent precursor to DHA, with an advantage of 6.4 IQ points (t = 6.35, p < .001) in children breastfed over those fed milk formula without LC-PUFA supplementation (Caspi et al., 2007). Unfortunately, the Western diet in general has shifted from a historic omega-3 to omega-6 ratio of approximately 1:1 to ratios upward of 1:25, largely in part due to a paradoxical recommendation to consume unsaturated fatty acids to diminish the risk of heart disease (Simopoulos, 2000). The change in n-3/n-6 ratios has correlated to an increased risk of inflammatory and cardiovascular diseases (Ghosh, Novak, & Innis, 2007).
The greater proportion of women in American and Canadian society consume much less DHA than the suggested 300 mg/day, although fetal demand is high, evidenced by the preference for DHA over other LC-PUFAs in the basal membrane of the placenta (Campbell, Gordon, & Dutta-Roy, 1996; Dutta-Roy, 2000), without concurrent increase in mothers’ dietary selection for foods high in DHA concentration. Inadequate supply to the fetus may lead to irreversible neurological damage (Szajewska, Hovarth, & Koletzko, 2006). Women with gestational diabetes often possess lower concentrations of DHA in breast milk when compared with those without (Lammi-Keefe, Rozowski, Parodi, Sobrevia, & Foncea, 2008). Typical American diets result in reduced DHA in breast milk compared with the foreign equivalent (Figure 3), due to consumption of foods heavy in AA and its precursors, which contributes to diminished DHA in the developing brain and hinders neurite outgrowth (Ikemoto et al., 1997; Kan et al., 2007).
Excessive supplementation of DHA may also disrupt neuron membrane permeability and enzyme function and, without adequate antioxidants, may create an accumulation of lipid peroxides (Figure 1:8; Horrocks & Yeo, 1999) that can induce a runaway inflammatory insult to neuronal tissues. Neuroprostanes are generated within the brain through enzyme-independent reactions due to oxidative stress leading to apoptosis (Mukherjee et al., 2004). Tests of visual acuity, measured by the Teller Acuity Card procedure, and both transient visual evoked potential latency and amplitude were lower in children receiving DHA-supplemented breast milk at 8 months of age compared with control (Jensen et al., 2005). At 12 months of age, neurodevelopment outcomes of those children receiving DHA supplemented only breast milk were assessed using the development quotient from the Clinical Adaptive Test (CAT DQ) and the development quotient from the Clinical Linguistic and Auditory Milestone Scale (CLAMS DQ). Although significant differences were not detected in either test, that in both instances, children receiving the DHA supplemented only breast milk scored lower than control is interesting (Jensen et al., 2005).
Similar negative findings came from two studies of attention-deficit hyperactivity disorder (ADHD) children (Hirayama, Hamazaki, & Terasawa, 2004; Voigt et al., 2001) where treatment with DHA alone was associated with results significantly lower on most measures than placebo. In addition to the possible generation of lipid peroxides, this article asserts that excessive DHA supplementation of infant formula, in combination with antioxidants during sensitive developmental stages, may result in premature neuronal hardwiring due to its powerful antiapoptotic neuroprotection. Such circumstances may contribute to autistogenesis in genetically or environmentally predisposed infants. This represents a case where neuroprotection may inhibit apoptosis in some regions necessary for appropriate developmental changes.
Studies of infant development linked to breast or bottle feeding, with or without LC-PUFA supplementation, are rife with challenges. Following the 1960s, breastfeeding increased but declined during the 1980s, increased during the 1990s, and appears to have peaked in 2002 (Abbott Laboratories [as cited in KellyMom, 2011]). With regard to pervasive developmental disorders, one study detected no significant difference in breastfeeding between 50 children with pervasive developmental disorders compared with 50 control children (Burd et al., 1988). A follow-up study (Schultz et al., 2006) reports that autistic children were significantly less breastfed and less likely to receive formula supplemented with DHA/AA than typically developing children. Less IgA and limited LC-PUFAs contributes to immune system dysfunction and represents potentiating factors in the development of autism (Schultz et al., 2006). However, it is an unfortunate fact that adequate data does exist for orphanage (Hunt, 1941; Spitz, 1945) and foundling home children, receiving only essential nutrients and exposed to social isolation and abuse, which demonstrate minimum behavioral characteristics diagnostic of autism (Rutter et al., 1999; Smith et al., 2008; Tustin, 1991, 1994).
Due to problematic designs and contradictory results, the final conclusion from two critical reviews (Jain, Concato, & Leventhal, 2002; Koo, 2003) indicate no evidence regarding the comparative effects of breastfeeding on intelligence and vocabulary linked to mothers’ orally supplemented breast milk or supplemented infant milk formula. Confounds for studies reviewed include cultural food preferences and availability, formally tested intelligence and traditional schooling of the parents, age and experience of the mother, sex and birth order of the infant, power and duration of the test, lack of rigorous dietary control of the mother, socioeconomic status, and purchasing power required for supplemented milk formula.
Diets containing various DHA to AA ratios were made available to rats. At ratios between 0.56 and 1.77, blood pressure is diminished through suppressed platelet aggregation without diminished platelet count (Yamada et al., 1997) in vitro. In two more recent studies, human DHA/AA ratios within the cerebral cortex were measured at no less than 1.25 ± 0.06 and not more than 1.90 ± 0.07, and within the hippocampus of not less than 1.20 ± 0.05 and not more than 1.67 ± 0.09 (Hashimoto et al., 2002; Hashimoto et al., 2005). Ratios of this type were associated with diminished neurological damage due to oxidative stress, diminishment of memory errors, improved spatial cognition, and reduced forebrain ischemia-induced cognitive deficiencies (Hashimoto et al., 2002; Hashimoto et al., 2005).
Rapoport, Rao, and Igarashi (2007) provided an analysis of several studies directly assessing gray matter phospholipids and indicate the DHA/AA ratio of healthy brain tissues of approximately 1.35. Dietary ratios of DHA/AA below 0.56 (an AA/DHA ratio of 1.80) contribute to increased lipid peroxide levels, partly because the increased AA reduces the presence of DHA by decreasing its synthesis, and through physical replacement within membranes (Hashimoto et al., 2002). Abbott, the makers of Similac, as well as Mead Johnson, the makers of Enfamil, offer commercially prepared infant formula with DHA to AA since 2002 with ratios of approximately 0.37 and 0.5, respectively (Similac Consumer Relations, personal communication, May 20, 2010; Enfamil Product Information, personal communication, May 20, 2010). Abbott representatives state that Similac established their DHA to AA ratio to replicate American women’s breast milk, which most closely resembles a study (Jensen, Maude, Anderson, & Heird, 2000), indicating a DHA/AA ratio lower than that characteristic of typical neurons (Kan et al., 2007), and 0.74 lower than the lowest of the top 10 countries (Figure 3) for DHA/AA in breast milk worldwide (Brenna et al., 2007).
Breast milk deficiency is undoubtedly due to diet as current American EPA and DHA intake is estimated at approximately 10% of optimal (Nestel, 1987; Neuringer, Anderson, & Connor, 1988). Abbott’s PediaSure and Mead Johnson’s Enfagrow Premium (vanilla and chocolate) contain DHA without AA (Similac Consumer Relations and Enfamil Product Information, personal communication, May 20, 2010). The Hain Celestial Group, Inc., makers of Earth’s Best infant formulas, as well as Gerber, Nature’s One, O for Baby, Nutramigen, and Vermont Organics, also use a DHA to AA ratio of 0.5 (Hain Celestial Group, Inc. Consumer Relations, personal communication, March 23, 2011).
Makrides et al. (2009) demonstrated that high-dose DHA (1% total fatty acids) administered through preterm infant milk formula did improve Bayley Mental Development (MDI) scores at 18 months corrected age in female infants only. This raises an important point and leads to understanding another aspect of autism’s overwhelming gender bias. Burdge, Jones, and Wootton (2002) provided controlled diets to males of 27 to 40 years and median body mass index (BMI) containing C13-labeled ALN at dosage 16 times higher than previous studies (Vermunt, Mensink, Simonis, & Hornstra, 1999) to assess conversion to DHA. Results indicate that approximately one third of ALN is used for energy during the first 24 hr following ingestion, and although capable of converting ALN to EPA and DPA to a limited extent, DHA synthesis in males was not sufficient to detect above background (Burdge et al., 2002). As estradiol also enhances learning and provides neuroprotection against damage due to ischemia (Zhang et al., 2004), the link between estradiol, DHA, and gender disparity is clear.
DPA is often overlooked in the literature regarding the link between lipid metabolism and autism, and this needs correction. DPA accumulates in the brain when DHA levels are low, and this occurs especially during fetal and infant development, maintaining the synthesis of phosphotidylcholine (Galli, Trzeciak, & Paoletti, 1971; Kim, Bigelow, & Kevala, 2004). DPA is limited in its ability to translocate Akt, and so does not well support neuronal survival following insult or injury (Garcia, Ward, Ma, Salem, & Kim, 1998; Hamilton, Greiner, Salem, & Kim, 2000). However, with three pathways for potential generation, DPA represents the primary route to growth and plasticity in response to DHA deficiency as both LNA and ALA pathways can produce DPA (Figure 2:9, 10, 11).
In a follow-up study, female conversion of ALN into EPA, n-3 DPA, and DHA was significantly greater than men from all previous studies (Burdge et al., 2002) and found greatest conversion of ALN to DHA among those participants concurrently using an estradiol-based birth control pill. The difference in ALN to DHA conversion between males and females is due to estrogen up-regulation (Figure 1:1; Figure 4:1), which explains the 25% variation in DHA detected during pregnancy (Burdge & Wootton, 2002). A more recent study (Giltay, Gooren, Toorians, Katan, & Zock, 2004) found ALN to DHA conversion 15% higher in women than men, and that among women, those taking oral 17β-estradiol contraceptives had 10% higher DHA concentrations than those not on oral contraceptives. Administrations of both testosterone and the aromatase (Figure 4:1; Figure 6:6) inhibitor anastrozole, which blocks the conversion of testosterone to estradiol, decreased DHA conversion (Giltay et al., 2004).
The Genetics of Lipid Metabolism and Sex Hormones in the Brain
The issue of autism genotypes and subphenotypes is a principal concern. Many signal linkages are found on Chromosomes 2, 3, 7, 8, 11, 15, 16, 17, and 19, depending on the test of psychological impairment used to diagnose the disorder (Liu, Paterson, & Szatman, & The Autistic Genome Project Consortium, 2008), excluding disorders of clear chromosomal etiology such as Rett syndrome and fragile X syndrome. Analysis of site-specific changes to the long arm of Chromosome 15 links it to the disorganized subtype of schizophrenia (Freedman et al., 1997), language delays, IQ = 70, P50 sensory gating disorder, and the α7-nicotinic cholinergic receptor (CHRNA7) that modulates activation of localized neuronal circuits in the human cerebral cortex (Albuquerque, Pereira, Eisenberg, Maelicke, & Alkodon, 2000).
In the brain, proinflammatory eicosanoids are produced through the action of cyclooxygenation, lipoxygenation, and cytochrome P450-dependent monoxygenation of DHA and AA. The metabolites strongly impinge on synaptic function and represent potential biomarkers for neurodegenerative states (Bazan, 1971; Montuschi et al., 2004; Phillis, Horrocks, & Farooqui, 2006) such as early stages of some autism subphenotypes. Up- or down-regulation of P450-dependent monoxygenation due to mutation, methylation, or in response to extreme dietary LC-PUFA imbalance provokes catastrophic risk to neuronal form and function. Cytochrome P450 represents a small yet powerful family of enzymes, and evidence (Fatemi, Reutiman, Folsom, & Sidwell, 2008; Gervasini, Carrillo, & Benitez, 2004) suggests that members from this group are implicated in disorders on the autistic spectrum. This is because faulty cytochrome P450 activity results in LC-PUFA metabolism similar to peroxisomal dysfunction (Kane & Kane, 1997).
The CYP19A1 gene (Figure 4:1), located at 15q21.2 (Chen et al., 1988; Simpson et al., 1994), codes for cytochrome P450aromatase (P450arom) that converts testosterone into neuroprotective estradiol (Zhang et al., 2004). Estradiol is the most ubiquitous and biologically active endogenous steroid, and has overwhelming influence on the developing brain. Estradiol functions in concert with estrogen receptors at peak in the final trimester of pregnancy, gradually tapering off from the first few weeks of life postpartum, followed by a massive prepubertal surge (Figure 7) weeks later (Abramovich & Rowe, 1973; Fitch & Denenberg, 1998; Forest, Sizonenko, Cathiard, & Bertrand, 1974), and then stabilizing at adult levels following puberty. Testosterone from the male testes crosses the blood-brain barrier and is converted by aromatase into estradiol (Figure 6:8). which encourages axonal regeneration, promotes synaptogenesis, stimulates neurogenesis, improves survival rates secondary to insult or injury, and modulates dendritic spine formation (Quesada, Lee, & Micevych, 2009), including connections to olivary cells.
The neonatal testosterone surge in males begins postpartum (Abramovich & Rowe, 1973; Forest et al., 1974), just as maximal DHA levels fall, at the same time that female infants experience a surge in estradiol (Figure 7; Fitch & Denenberg, 1998). In both sexes, the increase occurs in conjunction with the pituitary surge (Main, Schmidt, & Skakkebæk, 2000) that initiates rapid skeletal growth. Under healthy conditions, the prepubertal surge results in stabile neuronal development in males through the neuroprotective effects of testosterone (Figure 6:9) converted into estradiol (Deroo & Korach, 2006; Humphrey, 1998; Klein, Baron, Colli, McDonnell, & Cutler, 1994; Quesada, Lee, & Micevych, 2009).
Evidence suggests that estradiol modulates both apoptosis and neurogenesis in a region and context-specific manner (Arai, Sekine, & Murakami, 1996; Fukudome et al., 2003; Ma et al., 1993; Rasmussen, Torres-Aleman, McLusky, Naftolin, & Robbins, 1990). When conversion of testosterone to estradiol does not occur appropriately (Figure 6:10), androgens accumulate and obstruct conversion of homocysteine into the powerful brain antioxidant, glutathione (Vrbikova et al., 2003). Estrada, Varshney, and Ehrlich (2006) demonstrated that chronically elevated levels of testosterone trigger apoptosis in neuroblastoma cells and increased concentration in interneural calcium levels (Figure 1:6), which induce aberrant neuronal migration. Excessive androgen levels also increase generation of free radicals, enhanced lipid peroxidation of DHA and AA into toxic inflammatory metabolites, and greater inflammatory cytokine activity (Blaylock, 2009). Conversely, aromatization of normal levels of testosterone to estradiol improves verbal and spatial performance (Cherrier et al., 2005; Okkelova et al., 2003).
McCarthy (2008) found that a dose of estradiol administered to an immature brain requires several hours to alter dendritic formation, yet creates lasting and substantial neurological changes, where minutes suffice in adults but yield only temporary results. Similar to its influence on PCs, estradiol promotes the growth and development of axons within the ventromedial nucleus (VMN) of the hypothalamus of males and is mediated at least at the cellular level (McCarthy, 2008). It is helpful to remember how powerful the effects of female sex hormones are on circuits traversing the MNS, the orbital frontal cortex, the limbic system, the fusiform gyrus, and the superior temporal sulcus.
Excessive estradiol concentrations in females are also capable of impairing prosocial behavior, altering the effects of socially mediated reward/retribution processing, and may elicit functional dysregulation of emotional face recognition (Protopopescu et al., 2007; Rubinow, Smith, Schenkel, Schmidt, & Dancer, 2007), each of which represents characters found on the autism spectrum. The biology of premenstrual dysphoric disorder in females may provide further clues to the pathology of autism, particularly as disorders on the autism spectrum often present with impaired emotional face recognition (Wright et al., 2008) associated with diminished cell size, number, and activity in the fusiform gyrus (Bolte et al., 2006; Hall, Szechtman, & Nahmias, 2003; van Kooten et al., 2008). Furthermore, Ingudomnukul, Baron-Cohen, Wheelwright, and Knickmeyer (2007) indicated that women with autism spectrum conditions (ASC) report increased testosterone-related hirsutism, bi- or asexuality and “tomboyism,” menstrual cycle irregularities, severe acne, and family histories of ovarian, uterine, and prostatic neoplasia.
To a limited extent, estradiol is synthesized de novo differentially across varying regions of the cortex, and the neonatal hippocampus generates up to 50%, yet cytochrome P450arom activity only begins postnatally (MacLusky, Walters, Clark, & Toran-Allerand, 1994). McCarthy (2008) suggested that differing aspects of the developing brain are immune to the rapid and powerful influence of estradiol at different stages to prevent permanent aberrant neuronal hardwiring due to its powerful antiapoptotic neuroprotection. This is well in line with Kritzer (2006) who demonstrated that cortical development is regulated postnatally by estradiol via the alpha and beta estrogen receptor subtypes at different times, in differing patterns, and on different cortical cells.
That long-term potentiation is enhanced by estradiol in the forebrain (Mukai et al., 2007; Woolley, 2007) is important considering the many regions of the brain developing through sensitive periods. Furthermore, these studies support others indicating that estradiol modulates working memory (Sinopoli, Floresco, & Galea, 2006), object recognition and spatial memory (Luine, Jacome, & Maclusky, 2003), male aggression (Trainor, Lin, Finy, Rowland, & Nelson, 2007), and thus alters the dynamics of behavior specifically and information processing more generally. The MNS is perfused throughout the cortex for rapid information processing concerning perceived behavior of both the observed and the observer (Mukamel et al., 2010). This work asserts that estradiol will modulate developing aspects of this system and that the MNS (Iacoboni & Dapretto, 2006) must also represent the primary functional component of procedural memory, as well as an important neurological correlate to autistogenesis.
Estradiol also modulates oxytocin receptor (OXTR) sites (Nissenson, Flouret, & Hechter, 1978), which are known deficient in autism (Gregory et al., 2009) and are linked to social cognition (Gill, 2010). This understanding provides a well-founded theoretical explanation for why OXTR is greatly diminished in autism and yet RNA expression for OXTR remains normal (Tansey et al., 2010). Because estradiol also regulates conversion of ALN to DHA, the aromatase gene appears a strong candidate for consideration as the genetic basis for both gender disparity and environmental triggers in autistogenesis.
Recent Findings on Genetic Regulatory Mechanisms
Although the CYP19A1 gene (Fujita et al., 2010) is approximately 130,542 (130-kb) base pairs long, only approximately 30 kb encodes for aromatase, which converts testosterone to estradiol. The remainder codes for nine regulatory units (Figure 4) that impart tissue specificity: ovary/breast cancer/endometriosis, adipose/breast cancer, bone, Placenta Minor 1, brain, fetal tissues, skin/adipose, Placenta Minor 2, placenta major (Sebastian & Bulun, 2001). In each instance, the tissue-specific exon splices onto the aromatase coding region, thereby providing tissue-specific expression (Sebastian & Bulun, 2001). The great length of this gene provides enhanced opportunity for transcript-level regulation, increased chance for mutation, and multiple opportunities for failure at the common splice site. This work asserts that 15q21.2 (Fujita et al., 2010) is likely responsible for many testosterone-linked behaviors described by Ingudomnukul et al. (2007). Furthermore, epigenetic methylation may alter expression, and copy-number alterations within the 15q21.2 (Figure 5) may induce a reading frame shift in neighboring 15q23, which is implicated in previous studies (Liu et al., 2008) and demonstrates an estrogen-regulated phenotypic relationship with dyslexia (Massinen et al., 2009).
Another genetic source of dysfunction is pseudogene expression and micro-RNA (miRNA)–linked regulation. Pseudogenes are defective genomic loci accumulated through phylogenic development once interpreted as nonsense DNA because they acquire silent mutations that impede successful protein translation (Lee, 2003). Pseudogenes are well conserved and, through duplication and retrotransposition, have come to represent perhaps as much as half of the transcribed genome (Harrison, Zheng, Zhang, Carriero, & Gerstein, 2005). Because cells only employ those aspects of the genome necessary for functioning as part of their organizing tissue, transcription of these genetic elements is highly tissue specific, although only a very small sample has been functionally characterized (Poliseno et al., 2010).
In 2009, Bartel described a type of noncoding RNA sequences, miRNA, as biologically active regulators of transcription that function by binding to mRNA transcripts and impair translation. Pseudogene and competitive endogenous RNA (ceRNA) transcription may also result in the competitive inhibition of mRNA resulting in tissue pathogenesis (Poliseno et al., 2010). The significance of these findings cannot be overemphasized as some of these lesser known candidates contributing to autistogenesis may not demonstrate a correlative signal on current linkage analysis (Liu et al., 2008), and all may impinge on CYP19A1 at 15q21.2.
Discussion
Autism is a pervasive developmental disorder that includes both a qualitative and quantitative impairment of social interaction, cyclic CNS activity producing repetitive physical and cognitive behaviors, deficits in communication skills of all types, and an aversion for extremes in sensory novelty linked to a preference for environmental sameness. Sensitive periods exist as a consequence of plasticity during infant brain development due to overwhelming synaptic remodeling of gray matter connections in different regions, at different rates, and at different times whereas other regions experience advancing myelination. The result is specific adaptation of pathways contributing to aspects of anthropoid cognition such as perception, attention, learning, memory, symbolic representation, and the scaffolding on which is built imitation and theory of mind.
Injury or insult to sensitive brain regions results in cortical dysgenesis and retention of unsuitable interconnections. Any cellular disruption may induce neuroinflammation leading to reduction in perceptual, intellectual, language, and learning capacities, and diminished self-regulation, and distortion of emotional and social operations result. Important dietary antioxidants are metabolized with female gender bias and are consumed at trace levels only in the modern Western diet (Sen, Khanna, & Roy, 2006). Furthermore, only females demonstrate significant ability to synthesize DHA, to meet the demands of developing infants. Preterm and term infants can produce AA after 33 weeks gestation (Uauy, Mena, Wegher, Nieto, & Salem, 2000), which calls into question AA supplementation. There exists competition for enzymes (Figure 7) between n-3 and n-6 fatty acids (Brenner, 1974) so the relative proportions of each carry biological consequences. Adequate supplies of these LC-PUFAs, in the correct ratio, inhibit the neurodegenerative aspects of inflammation and suppress apoptosis while concurrently enhancing long-term potentiation.
Incorrect ratios or inadequate levels of these LC-PUFAs result in reduced neurogenesis, diminished neurite outgrowth, and increased susceptibility to a runaway neuroinflammatory cascade, which includes additional cell death and the damaging effects of neuronal inflammation such as brain swelling constrained within the skull. The correct n-3/n-6 LC-PUFA ratio will mitigate or ameliorate insult and injury due to environmental toxins, ischemia, oxidative stress, excess sex hormones, and the many pathways through the neuroinflammatory cascade. Incorrect dietary n-3/n-6 ratios can enhance or even induce neurodegenerative processes in some instances.
The correct ratio of estradiol to testosterone in preterm mothers, neonates, and developing infants is critical for neurogenesis, neuronal differentiation, neurite outgrowth, and enhances long-term potentiation. Furthermore, estradiol inhibits the neurodegenerative inflammatory response and suppresses apoptosis. Testosterone is converted into estradiol by cytochrome P450arom, and failure in this sequence of events leads to significantly lower estradiol and increased testosterone. Diminished conversion of testosterone into estradiol inhibits neurogenesis, reduced neurite outgrowth, and greater vulnerability to cytotoxic metabolites from neuronal inflammation. Without functioning aromatase enzymes, normal levels of testosterone become excessive, generating free radicals that transform DHA and AA into cytotoxic inflammation metabolites through increased lipid peroxidation.
Therefore, this article asserts that a spectrum of developmental disorders results from damage or impairment to one or more components within a system of overlapping spheres of influence: (a) the genetic mechanisms and metabolic processes of testosterone conversion to estradiol, (b) estradiol’s regulation of oxytocin and n-3 fatty acid metabolism, (c) the central role of fatty acid metabolism in neuroinflammation, and (d) the inflammatory response initiated by environmental stressors (Figure 5). The CYP19A1 gene represents the center of a web of events responsible for gender disparity. Estradiol, EPA, DPA, DHA, and NPD1 mediate neuronal development and specialization while safeguarding against cascading neurodegenerative events when interconnectivity is most susceptible. Dyslexia, attention-deficit disorder (ADD)/ADHD, certain types of language impairment, and varying degrees of antisocial behaviors are grossly overrepresented in males (Auyeung et al., 2009). These developmental challenges are responsive to DHA supplementation (Richardson, 2006), and the systems theory of autistogenesis provides a powerful framework for explaining primary aspects of their respective etiologies, including nearly a dozen mechanisms responsible for gender bias.
Environmental influences have powerful sway over sensitive aspects of the developing infant brain and can induce hard-wired changes in the maturing primate brain attempting to adapt to an inhospitable environment. Any strongly acute or chronic environmental stress will initiate the neuroinflammatory cascade, as in the cytotoxic effects of cortisol or testosterone, but social and emotional stress can also cause the initiation of DHA and AA cascades through neurotransmission through PLA21 coupled receptors (Rapoport, Rao, & Igarashi, 2007). Toxic metal exposure, overwhelming allergic reactions, the effects of bacterial and viral infection, metabolic dysfunction and dietary inadequacies, and direct injury can lead to unsuitable maturation through myelination of aberrant connections following synaptic pruning under the weight of neuroinflammatory processes. Neuroinflammation results in an expanding sphere of physiologic debridement, whereas the remaining cells experience aberrant myelination leading to developmental disorder.
Where most young children can tolerate a degree of stress for a certain period of time, others with deficient P450arom expression or function may not develop healthy connections during sensitive periods. An infant with competent aromatase function may develop aberrant connections if exposed to reasonably normal levels of stress if concurrently oversupplemented with AA that promotes the feed-forward neuroinflammatory response. Infants receiving excessive nutritional supplementation of DHA alone may not experience appropriate cell death, and so lose some remodeling in domains where new connections must form, while other areas wither under DHA’s potential for proinflammatory influence.
Infant males moving into the testosterone surge incapable of aromatase conversion will not possess neuroprotective capacity to protect against testosterone toxicity (Figure 7) or environmental stress as DHA concentrations within the brain fall after parturition. Only healthy breast milk or correctly formulated infant milk replacement may provide an adequate DHA/AA ratio. Infants with faulty P450arom genetics may grow normally, but overwhelming stress during sensitive stages of development may induce regression as neuroinflammation results in physiological debridement. Circumstances such as these explain how seemingly similar male children exposed to similar stressors may suddenly depart along very different developmental trajectories.
Implications and New Directions for Study
The implications that arise from the assimilation of such diverse research are numerous and staggering. Mutation, intrauterine epigenetic methylation (Kaplan, Evans, & Monk, 2008; Roth & Sweatt, 2011; Wright & Enlow, 2008) or transcript-level regulation of CYP19A1 at the aromatase coding region, the common splice, or any of nine tissue-specific regulatory regions can disrupt conversion of testosterone into estradiol. This is cause for concern during the prepubertal testosterone surge that peaks as critical periods in developmental sensitivity begin (Ajlouni, Daoud, Ajlouni, & Ajlouni, 2010; Alexander, Wilcox, & Farmer, 2009; Hadziselimovic, Emmons, & Buser, 2004). Between testosterone production, estradiol synthesis, and regulation of DHA metabolism, there are many opportunities for genetic and metabolic failure.
Infant milk formula supplemented with a DHA/AA ratio determined previously as potentially detrimental could have potentiated circumstances in some children now on the autism spectrum that may never have been. A new line of study must examine whether EPA, DPA, or DHA, alone or together, in combination with antioxidants, would not prove an effective intervention. Whereas both DPA and DHA enhance neuronal elongation and neurite outgrowth, only DHA supports growth under dietary disturbance or environmental stress. Although the neuroprotective properties of DHA are impressive, apoptosis makes way for new connections, and myelination may hinder plasticity. It therefore seems that DPA, which promotes growth while maintaining plasticity, could prove a better choice for other treatment needs.
The three overlapping spheres of primary influence in autistogenesis are clear: the genetics supporting aromatization of testosterone into estradiol, LC-PUFA metabolism as modulated by estradiol, and the thousand natural shocks that flesh is heir to. Gender disparity is important to understanding the etiology of autism and subphenotypes, including many learning disabilities overrepresented in the male population. New studies must examine the effects of dysregulation of an already complex CYP19A1. This gene controls or influences placental DHA absorption and mammary gland DHA production, and the developing infant’s levels of testosterone and estradiol. CYP19A1 also regulates oxytocin, MET response, antioxidant metabolism, and modulates several aspects of the inflammatory response. Efforts must also seek to determine the effects of at least 13 known CYP19A1 alleles (Online Mendelian Inheritance in Man, 2011) and the gene’s multiple inherent potentials for failure. Such studies will necessarily include consideration of epigenetic methylation, pseudogenes, miRNA, ceRNA, and any form of posttranslational modification because most if not all of these may go undetected in traditional linkage assays. Furthermore, as each of the above represents a possible gear in the autistogenesis mechanism, it becomes clear that a bioassay for early detection will include polymerase chain reaction (PCR) screening for CYP19A1 alleles, pseudogenes, and transcription-level regulators.
Current attempts at linkage analysis demonstrate a key misunderstanding because environmental stimulus can induce pervasive developmental disorders. Correctly functioning testosterone aromatization remains a potential causative factor if environmentally predisposed, especially with incorrect DHA/AA supplementation, and this explains why CYP19A1 may not produce a linkage signal with logarithm of odds scores as high as 3.6; a well-functioning gene can still predispose the disorder (Figure 5). Testosterone overexpressors are not genetically well characterized, and research must determine if there is a significant presence of developmental disorders in families possessing such a trait. As the male does not produce significant levels of DHA, and precisely because both males and females do produce AA, excessive AA supplementation can shift into metabolic pathways toxic for neuronal growth and development, producing a runaway effect. The inflammatory cascade can also result in nonenzymatic oxidation of DHA when present in excess, which generates yet more highly reactive toxic metabolites. Researchers must determine the effects of repeatedly acute and chronic stress on neonatal development concurrent with varying DHA/AA ratios and concentrations to understand the ramifications of infant milk formula based on deficient models.
Studies are also needed to determine the exact means by which estradiol governs LC-PUFA metabolism in response to a variety of environmental stressors. As estradiol is powerfully neuroprotective, studies are needed to determine if this is due to a fundamental property of estradiol alone, or due to the close link with LC-PUFA metabolism. It remains a question whether estradiol shuttles 24:6n-3 through the peroxisome membrane, but it may be that estradiol insensitivity can occur as a result of the testosterone surge, something akin to the etiology of diabetes mellitus type 2. This is an interesting hypothesis as adrenoleukodystrophy (ALD), a peroxisomal disorder leading to profound neurodegeneration and sometimes death, is a male-only disease and some cases may result not from inadequate conversion of LC-PUFAs but rather a deficit due to estradiol insensitivity.
For every step in the genetic, metabolic, and environmental pathway to autistogenesis, there is an opportunity for early detection, treatment, and intervention. Neuronal maturation occurs throughout the brain in predictable fashion and these events provide a roadmap with mileposts to guide intervention. The younger the child is without competent aromatase expression, or with inappropriate dietary DHA/AA ratios, confronted by any neuroinflammatory event, the greater the chance for damage to sensitive domains. Likewise, the sooner that socially engaging and neurologically appropriate intervention is targeted to those specific brain regions, the greater chance for improvement. Therapy must include activity that provides neurodevelopmentally appropriate stimulation targeting areas likely damaged, such as the Floortime® model, so that remaining plasticity may remodel faulty interconnections. Neurologists employing modern scanning and imaging techniques might determine cortical areas with aberrant electrical activity, or inadequate BOLD response, and provide neuropsychologists with a plan for therapists to develop brain region–specific intervention and suitable dietary LC-PUFA supplementation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.