
Abstract
The current surgical approach for patients with a single ventricle is the culmination of decades of dedicated research and groundbreaking innovation. From 1971 to the present day, a series of incremental advancements have significantly extended the life expectancy of these patients. Since the very beginning, Dr Guillermo Kreutzer and his team have pioneered different techniques with the ultimate goal of improving outcomes for these individuals. This is, narrated by him, the story of how it all began.
My name is Guillermo Kreutzer and I have a tale to tell. It is a story from a long time ago, yet its impact resonates strongly in our present time. While we now find many things self-evident, in those days, everything was cloaked in darkness. I invite you to join me as we traverse the transition from night to day, uncovering the origins of this transformative journey. As the narrator, I present to you the chronicle of how it all commenced.
Pediatric Cardiology in Argentina was pioneered by Rodolfo Kreutzer, my father (Figure 1A). He joined the pediatric team at the Hospital de Niños “Ricardo Gutiérrez” in Buenos Aires in 1921, where he quickly developed a deep interest in Pediatric Cardiology. His initial focus was on rheumatic fever, but after Robert E. Gross reported the first patent ductus arteriosus closure in 1939, he shifted his attention toward congenital heart defects and the pursuit of accurate diagnosis. In 1940, following the groundbreaking work of Agustín Castellanos in Cuba, he successfully performed the third-ever angiocardiography (Figure 1B), a monumental achievement in the field of congenital heart diseases.
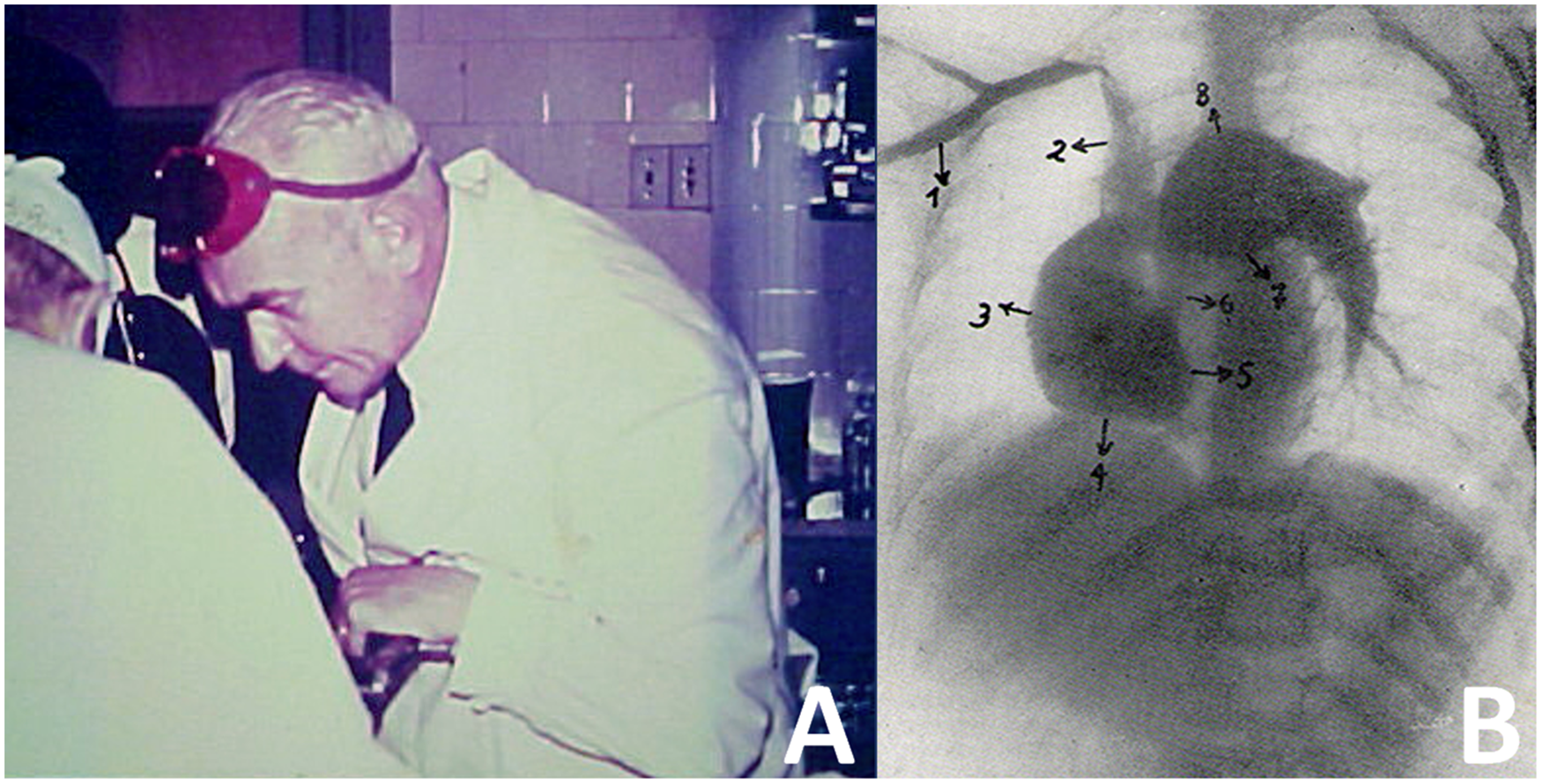
(A) Rodolfo Kreutzer at the Hospital de Niños “Ricardo Gutiérrez” in Buenos Aires; (B) original picture of the right angiogram in left anterior oblique view of a patient with pulmonary stenosis and patent ductus arteriosus (1: right subclavian vein; 2: superior vena cava; 3: right atrium; 4: right ventricle; 5: interventricular septum; 6: narrowed infundibulum; 7: main pulmonary artery; and 8: left pulmonary artery).
In 1961, I embarked on my Pediatrics residency with the goal of following my father's footsteps and becoming a Pediatric Cardiologist. In October of that same year, the Argentine Society of Cardiology organized a Congress, and they invited Professor Zerbini as a guest speaker. My father was in charge of the Pediatrics section of the Congress and extended an invitation to Professor Zerbini and Dr Brea to have dinner at our home. Dr Brea, in addition to operating on patients with congenital heart disease from the Children's Hospital, also served as the Dean of the Faculty of Medicine at the University of Buenos Aires. During a lull in our conversation, I gathered the courage to ask Dr Brea about the possibility of securing a scholarship from the University to pursue Pediatric Cardiology in London under the guidance of Paul Wood. The next day, during another meeting, Professor Zerbini approached my father and proposed having a chat with me to encourage me to consider training in Cardiovascular Surgery in Sao Paulo. My father's immediate reaction was, “Billy, a surgeon? Impossible!” However, Professor Zerbini insisted. The following day, during my interview, I expressed my disinterest in surgery and admitted that I had never witnessed an operation before. In response, he said, “If you've never been in an operating room, how can you be sure you won't like it?” He then extended an invitation for me to spend one month at his Cardiovascular Surgery Unit in Sao Paulo, with the option to stay for three years and complete my residency there. This is how, two years later, I performed my very first operation—a patent ductus arteriosus division—even though I had no prior experience in general or thoracic surgery.
In 1964, after my training in Sao Paulo with Professor Zerbini and Adib Jatene, we launched the Cardiac Surgery Program at the Hospital de Niños “Ricardo Gutiérrez” in Buenos Aires. In this prestigious institution of the public healthcare system, we inaugurated the first residency of Pediatric Cardiac Surgery in the country. In those early years, we made substantial progress in surgery in infants with complex lesions, including the use of irradiated homografts.
In July 1971, when I was 37 years old, a gravely ill three-year-old boy with an oxygen saturation level of 60% was admitted to our unit. He was diagnosed with tricuspid atresia type IB. Cardiac catheterization revealed an occluded right pulmonary artery as a consequence of a thrombosed prior Waterston shunt. The right pulmonary artery was not identifiable (Figure 2).
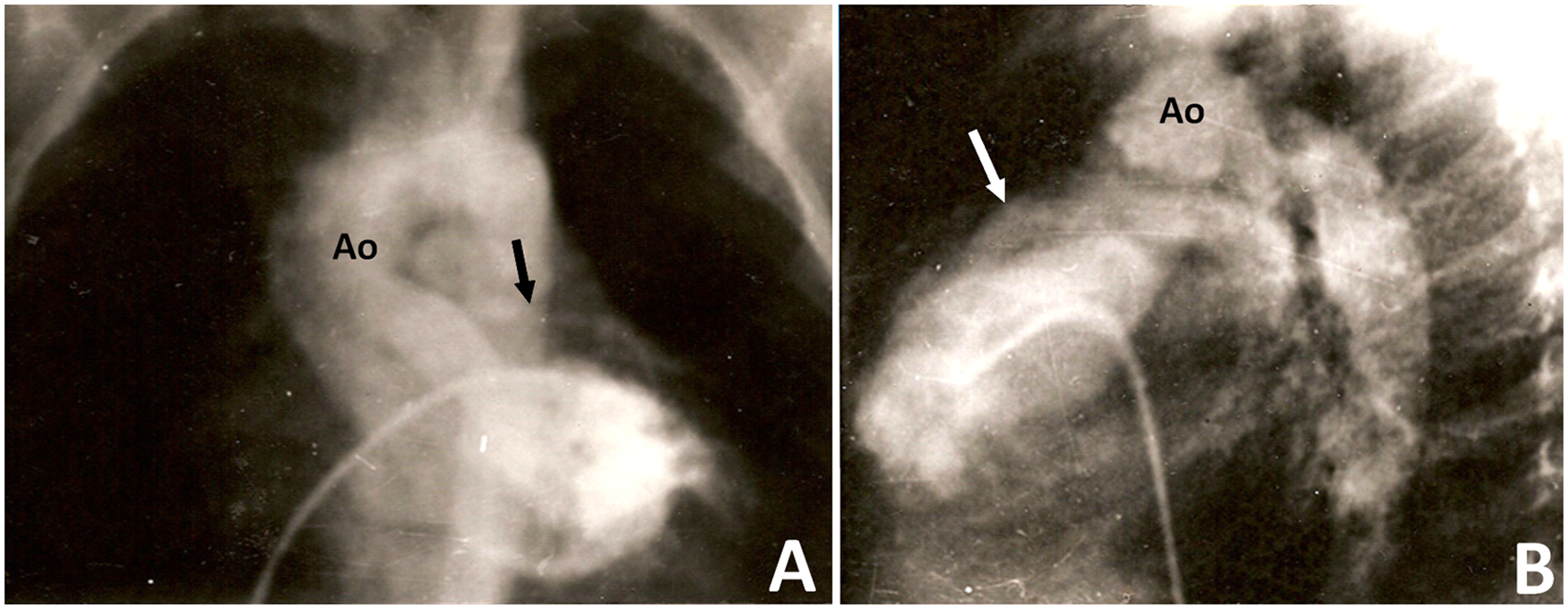
(A) No right pulmonary artery is visible, only the left (Ao: aorta; arrow: left pulmonary artery); (B) left anterior oblique view of the right ventricular outflow tract and left pulmonary artery (Ao: aorta; arrow: right ventricular outflow tract).
In those days, the available surgical options for patients like this were scarce. Performing a left Blalock-Taussig-Thomas shunt was deemed too risky due to the right pulmonary artery's lack of patency. It is important to note that the use of cardiopulmonary bypass for this procedure had not been described yet. The patient's mother implored me to do something for her dying child. Recognizing the gravity of the situation, we offered to attempt a completely novel surgical operation that we had devised but never attempted before. With her consent, on July 14, 1971, we performed an atriopulmonary anastomosis under cardiopulmonary bypass with intermittent aortic cross-clamping, placing a homograft between the right atrial appendage and the main pulmonary artery and leaving a 6-mm fenestration at the atrial septum (Figure 3A). We did not insert a valve in the inferior vena cava, nor did we perform a concomitant Glenn procedure.
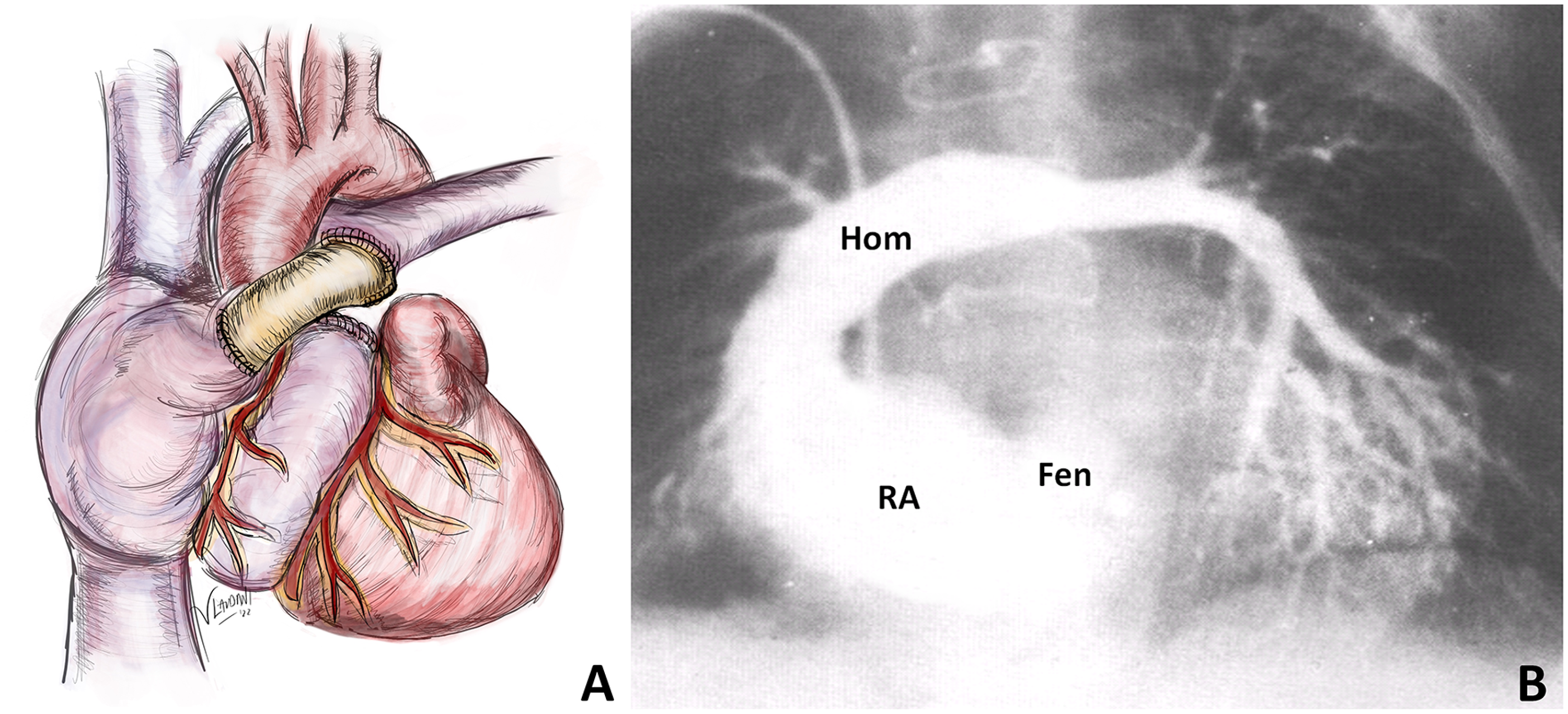
(A) Atriopulmonary anastomosis using a homograft; (B) postoperative angiogram showing the right atrium, the fenestration at the atrial level, the homograft and the left pulmonary artery, with minimal filling of the right pulmonary artery (Fen: fenestration; Hom: homograft; RA: right atrium).
Without any doubt, this was the first fenestrated total right heart bypass in history. At that time, access to the journals Annales de chirurgie thoracique et cardio vasculaire and Thorax was unavailable in Argentina, and therefore, we were not aware of Dr Fontan's publications, which had been published a few months earlier.1,2 The patient recovered uneventfully and was catheterized three weeks later (Figure 3B). The technique was presented in August 1971 at a meeting of the Argentinian Society of Cardiology.
In December 1971, another patient with tricuspid atresia type IB was admitted to our hospital. Dr Luis Becú, our cardiac morphologist, had already pointed out that in patients with this particular anatomic subtype, the pulmonary valve is usually normal. This crucial insight prompted us to adopt a unique approach for the atriopulmonary anastomosis procedure. We decided to perform the atriopulmonary anastomosis directly by harvesting the patient's own pulmonary artery root from the right ventricular outflow tract, following the technique pioneered by Donald Ross, and anastomosing it to the right atrium (RA). Our speculation was that this approach could facilitate growth and potentially prevent calcification over time. In 1973, both techniques were published in the Journal of Thoracic and Cardiovascular Surgery. 3 In that article, we highlighted that the total or partial closure of the foramen ovale was a topic of debate. While it served as a safety valve for the right atrium, it could also result in a certain degree of systemic desaturation.
Encouraged by our initial success, we made the decision to extend the procedure to other patients diagnosed with tricuspid atresia. During that era, a significant question arose: “Do we require a pump or simply a pathway?” Dr Fontan had proposed the concept of “ventriclization” of the right atrium. This approach involved the placement of a valve in the inferior vena cava and a homograft positioned between the right atrium and the left pulmonary artery, followed by complete closure of the atrial septum and a classic Glenn procedure. This method shared similarities with the concept of the one-and-a-half ventricle repair, but with the problem that the right atrium does not function as a ventricle.
Our approach was completely different. We opted for an atriopulmonary anastomosis, incorporating a 6-mm fenestration in the atrial septum. This decision was the result of extensive internal discussions involving all members of the Heart Team, including Dr Becú. When I presented him with the Fontan approach, Dr Becú expressed his skepticism, stating, “Billy, the right atrium will never become a ventricle.” Our concept also found support from Dr Alberto Rodríguez Coronel, the Head of the Interventional Cardiology Unit, who believed that diastolic relaxation of the left ventricle would serve as a driving force in the pulmonary circulation. As Dr Marshall Jacobs once told me, our team was the first to achieve a prescient and accurate understanding of this peculiar system.
By 1977, we had reached the understanding that the venous flow through the pathway was consistently continuous. As a result, we concluded that valves were redundant since they remained open at all times. In fact, their presence could even be detrimental, potentially leading to the formation of areas of stenosis within the pathway. Based on these findings, we made the decision to implement our third technical modification of the atriopulmonary anastomosis. This modification involved creating the widest possible anastomosis between the roof of the right atrial appendage and the transected main pulmonary artery, extending the incision further into the distal right pulmonary artery (Figure 4A). In cases of normally related great arteries, this entailed positioning the transected main pulmonary artery behind the aorta toward the right side. The proximal end of the main pulmonary artery (which was connected to the heart) was surgically closed and a deliberate fenestration was intentionally left in the atrial septum. In situations where double-inlet ventricle was present, we also proceeded to obliterate the right atrioventricular valve (if the left atrioventricular valve was normal). As a consequence, the resulting cross-sectional area at the site of the atriopulmonary anastomosis exceeded the combined cross-sectional area of both caval veins. This substantial increase in size, coupled with the inherent potential for growth, offered notable advantages to this procedure.
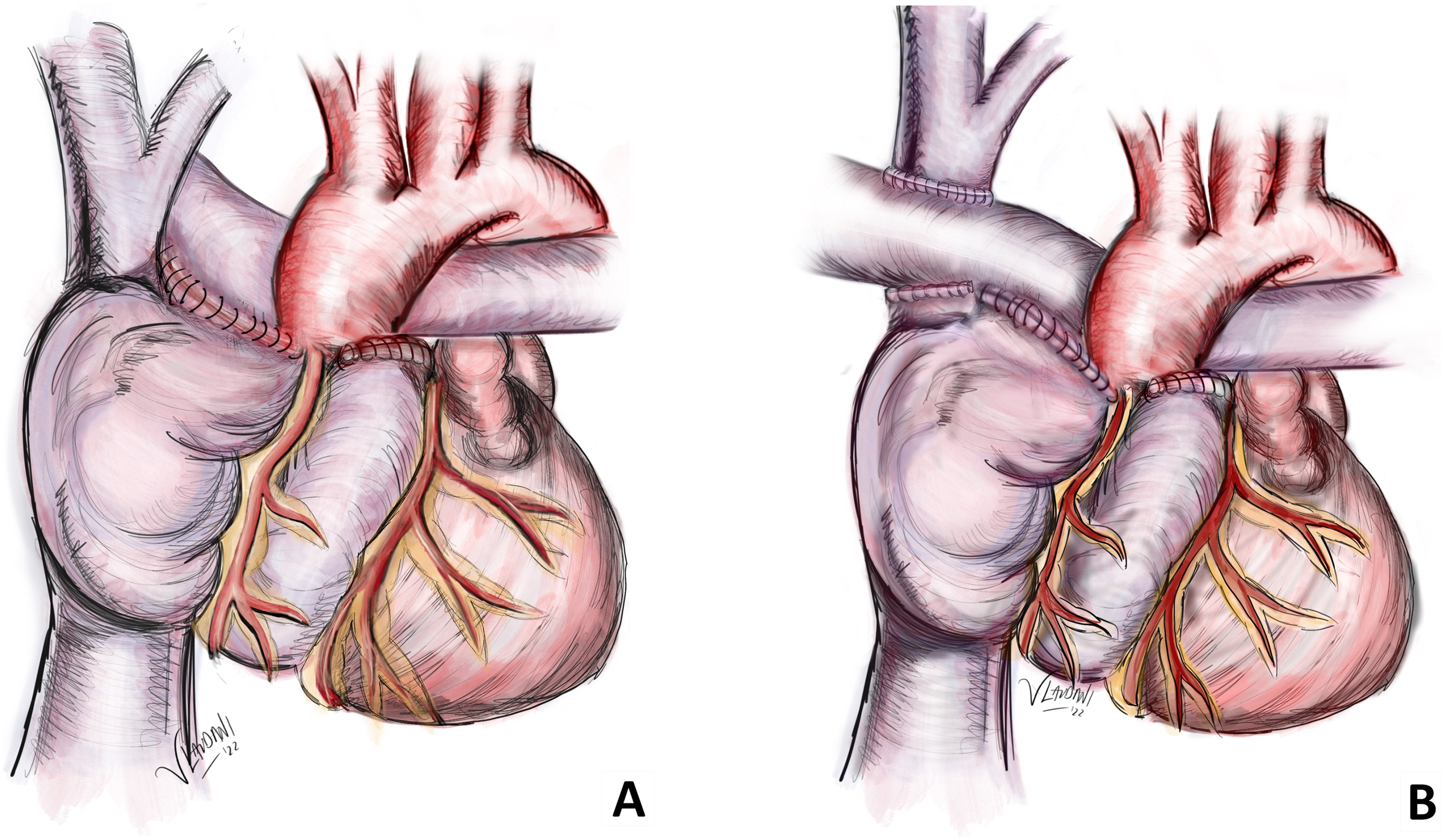
(A) Direct posterior atriopulmonary anastomosis; (B) direct posterior atriopulmonary anastomosis with a concomitant Glenn procedure.
Our experience was first presented in 1980 at the first World Congress of Pediatric Cardiology in London, and subsequently published in the Arquivos Brasileiros de Cardiologia in October 1981 4 and in the Journal Thoracic and Cardiovascular Surgery in 1982. 5 The technique gained global recognition as the preferred procedure in the late 1970s and early 1980s. However, after a few years, we started observing that the right atrium dilated in some patients, which subsequently resulted in the development of arrhythmia, thrombosis, and even compression of the right pulmonary veins. Consequently, in 1983, we introduced an additional component to the procedure, incorporating a bidirectional Glenn shunt (Figure 4B) to reduce blood flow coming to the right atrium.
In 1987, Puga and colleagues introduced a modification to the Fontan-Kreutzer operation aimed at preventing pulmonary venous obstruction in patients with single atrioventricular valve or left atrioventricular valve atresia. 6 This approach, later referred to as the “lateral tunnel technique,” involved transecting the superior vena cava and anastomosing the cardiac and cephalad ends separately to the confluence of the pulmonary artery. A patch inside the right atrium was used to create a separation between systemic and pulmonary venous drainages. In 1988, de Leval and colleagues published their experimental studies and early clinical experience with the lateral tunnel technique, involving 20 patients. 7 This strategy proved beneficial as it allowed for a significant portion of the right atrial chamber to remain at low pressure, thereby reducing the risk of arrhythmia. My concern with this approach was that the inferior vena cava is larger than the superior vena cava and could cause some pressure gradient in the pathway. However, the design of the tunnel minimized turbulence in comparison with the previous approaches, thereby reducing energy losses and lowering the risk of atrial thrombosis. Later on, the Boston group, led by Aldo Castaneda, revisited and validated the routine use of a fenestration, 8 just as we had previously described and implemented in all cases since 1971.
Around the same time, in 1988, Humes presented the experience of the Mayo Clinic in a series of 49 patients with asplenia or polysplenia syndromes. 9 In one patient, they attempted to replicate the experimental anastomoses of the inferior vena cava to the right pulmonary artery that had been reported in dogs by Nuland and colleagues in 1958, 10 but the patient died. Despite this setback, Puga and Marcelletti's group persisted with the approach and reported successful outcomes in seven patients with various forms of single ventricle. 11 This led to the widespread adoption of the extracardiac conduit technique by several teams globally, including our own.
All these continually evolving strategies have led to a dramatic increase in the survival and quality of life for patients with single ventricle heart disease. The atriopulmonary anastomosis served as a crucial milestone in this journey, demonstrating a survival rate of 70% at 35 years. 12 Several patients lived decades with this operation 13 and were then submitted to conversion or heart transplantation, that can be performed effectively with overall good long-term survival. 14 Therefore, it would be unfair to consider the Fontan-Kreutzer procedure as a failed strategy. 15 We must acknowledge that we are dealing with the most complex forms of congenital heart disease. Moreover, when comparing freedom from reinterventions and late survival outcomes, it is worth noting that some complex biventricular repairs (e.g. some of those including a Rastelli procedure) have shown worse results than the Fontan-Kreutzer procedure.16,17
Regrettably, not everything is rosy in our daily practice. Late failure remains a disappointing reality. However, it is crucial to recognize that suboptimal management is a key contributing factor, and it is our failure, not that of the Fontan-Kreutzer procedure. Late failure often presents with common iatrogenic issues such as pulmonary artery branch stenosis or absence, as well as conduit stenosis or distortion. From the beginning, subaortic stenosis should be diligently treated. It is imperative to prevent chronic volume overload caused by long-standing loose bands or large shunts. Phrenic nerve palsy is always a result of our actions and must be avoided. Perfect management of cardiopulmonary bypass, optimal organ protection, and technical excellence in constructing the Fontan-Kreutzer pathway should always be our top priority when dealing with these patients.
I have come a long way since the beginning of my career in 1961 to this day, at the age of 88. I have had the privilege of being part of a family historically devoted to the care of patients with congenital heart disease. My father, my brother Eduardo, and my children have all also made significant contributions in this field, impacting the lives of many. Together with my wife and grandchildren, they have accompanied me throughout this incredible journey. I have enjoyed every day of my professional life with mentors, colleagues, and friends. I am proud to have trained dozens of surgeons at a public hospital, where we performed more than 300 operations each year. These surgeons are the present and future of this specialty and I feel proud of the majority of them. I have humbly accepted many honors and recognition for my work (Figure 5). And above all, thanks to God, I am blessed to enjoy life today and be able to tell the tale.

(A) In 2011, during the “Symposium on univentricular heart: from A to Z” in Istanbul, I was honored to receive a recognition from Dr Christo Tchervenkov, the Executive Director of the World Society for Pediatric and Congenital Heart Surgery; (B) A special moment with my friend, Dr Francis Fontan.
Footnotes
Acknowledgments
To Dr Verónica Laudani, congenital heart surgeon, for her remarkable contribution with the figures.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.